Die fortschrittliche Deep-Learning-Software von HS Analysis ist in der Lage, eine Vielzahl von Krankheiten zu erkennen. Ein Beispiel ist die chronische myeloische Leukämie (CML), eine aggressive Krebsart, die in den blutbildenden Zellen des Knochenmarks beginnt und weiße Blutkörperchen und Knochenmark angreift, wodurch das Immunsystem geschwächt wird und sich die Krankheit im Körper ausbreitet. Spezialisten bei HS Analysis können mithilfe fortschrittlicher Deep-Learning-KI-Software CML fachgerecht diagnostizieren und Kliniken, Institutionen und Gesundheitseinrichtungen unterstützen.
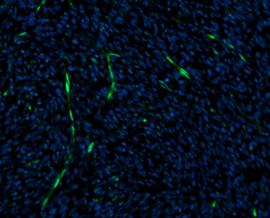
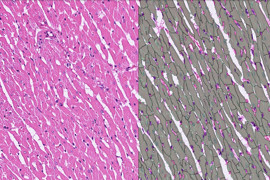
Gram-Färbung bei CML
In der Hämatologie werden verschiedene Färbemethoden verwendet, um Blutzellen und ihre spezifischen Komponenten sichtbar zu machen. Eine der am häufigsten verwendeten Färbemethoden ist die Gram-Färbung.
Die Gram-Färbetechnik wurde ursprünglich entwickelt, um Bakterien in der Mikrobiologie zu differenzieren. Sie basiert auf den Unterschieden in der Zellwandstruktur zwischen den Bakterienarten. Die Gram-Färbung kann jedoch auch in der Hämatologie verwendet werden, um bestimmte Zelltypen zu identifizieren.
Während des Gram-Färbeprozesses werden Blutzellen auf einem Objektträger fixiert und mit einer speziellen Färbemischung behandelt. Diese Mischung enthält typischerweise Kristallviolett, Lugolsche Lösung und Ethanol. Durch die Färbung behalten die Zellwände bestimmter Zelltypen die Farbe bei, während andere Zelltypen die Färbung während der nachfolgenden Behandlung verlieren.
Nach der Färbung werden die Zellen unter einem Mikroskop beobachtet. Typischerweise erscheinen Granulozyten wie Neutrophile oder Eosinophile nach der Gram-Färbung violett oder blau, während Lymphozyten und Monozyten ihre ursprüngliche Farbe verlieren und entfärbt werden.
Es ist jedoch wichtig zu beachten, dass die Gram-Färbung in der Hämatologie nicht routinemäßig angewendet wird. Andere Färbetechniken, wie die Giemsa-Färbung, werden häufiger verwendet, um Blutzellen zu färben und zu analysieren.
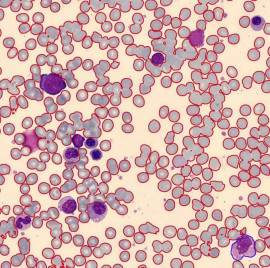
Die Gram-Färbung wird manchmal verwendet, um bei der Diagnose der chronischen myeloischen Leukämie (CML) zu helfen. CML ist eine Art von Blutkrebs, der durch eine Überproduktion unreifer weißer Blutkörperchen gekennzeichnet ist. Die Diagnose von CML erfordert spezifischere diagnostische Verfahren.
Die Diagnose von CML umfasst typischerweise eine Kombination aus körperlicher Untersuchung, Bluttests und bildgebenden Untersuchungen. Die wichtigste diagnostische Methode zur Bestätigung von CML besteht darin, das Blut unter einem Mikroskop zu untersuchen, um abnorme Blutzellen zu identifizieren.
Während der Untersuchung des Blutausstrichs werden spezifische Veränderungen in den Blutzellen beobachtet, die für CML charakteristisch sind. Dazu gehört das Vorhandensein des Philadelphia-Chromosoms, einer spezifischen genetischen Mutation, sowie eine erhöhte Anzahl unreifer weißer Blutkörperchen, insbesondere myeloischer Vorläuferzellen.
Weitere diagnostische Verfahren, die bei CML angewendet werden können, umfassen die Knochenmarkaspiration und Biopsie, bei der Gewebeproben aus dem Knochenmark entnommen und untersucht werden. Genetische Tests und molekulare Untersuchungen können ebenfalls durchgeführt werden, um das Vorhandensein spezifischer Genmutationen wie BCR-ABL1, die mit CML in Verbindung stehen, zu erkennen.
Es ist wichtig, dass die Diagnose und Behandlung von CML von erfahrenen Hämatologen durchgeführt wird, da es sich um eine komplexe Erkrankung handelt, die einen individuellen Ansatz erfordert.
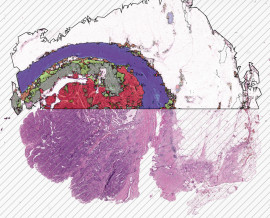
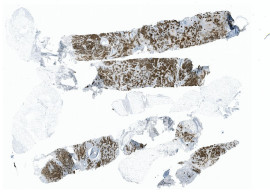
Knochenmarkdiagnose mit HSA KIT
Bei der chronischen myeloischen Leukämie (CML) können im Erscheinungsbild des Knochenmarks charakteristische Veränderungen auftreten, die auf die Krankheit hinweisen. Während einer Knochenmarkuntersuchung können mit der Immunhämatologie folgende Merkmale beobachtet werden:
- Erhöhte Zellzahl: Das Knochenmark bei CML ist typischerweise hyperzellulär, was bedeutet, dass im Vergleich zum Normalzustand eine erhöhte Anzahl von Zellen vorliegt. Dies ist auf die Überproduktion myeloischer Zellen, insbesondere Granulozyten (Neutrophile, Eosinophile und Basophile), zurückzuführen, die bei CML typischerweise erhöht sind.
- Linksverschiebung: Der Begriff „Linksverschiebung“ bezieht sich auf das Vorhandensein unreifer oder Vorläuferzellen im Knochenmark. Bei CML kann eine erhöhte Anzahl myeloischer Vorläuferzellen, sogenannte Myeloblasten, beobachtet werden. Diese Zellen sind im Knochenmark gesunder Personen normalerweise nicht in signifikanter Anzahl vorhanden.
- Philadelphia-Chromosom: Das Philadelphia-Chromosom, eine genetische Anomalie, die durch eine Translokation zwischen den Chromosomen 9 und 22 entsteht, ist ein Kennzeichen von CML. Diese Anomalie führt zur Bildung des BCR-ABL1-Fusionsgens. Das Vorhandensein des Philadelphia-Chromosoms kann durch zytogenetische Tests oder molekulare Techniken in den Knochenmarkzellen nachgewiesen werden.
- Erhöhte Megakaryozytenzahl: Megakaryozyten sind große Zellen, die für die Produktion von Blutplättchen verantwortlich sind. Bei CML ist häufig eine Zunahme der Anzahl und Größe der Megakaryozyten im Knochenmark zu beobachten.
HSA KIT unterstützt Sie dabei, diese Merkmale zusammen mit anderen klinischen und labortechnischen Befunden zu erkennen, die bei der Diagnose und Klassifizierung von CML helfen. Es ist wichtig zu beachten, dass das spezifische Erscheinungsbild des Knochenmarks von Person zu Person variieren kann und zusätzliche Tests, wie genetische Analysen oder molekulare Tests, für eine definitive Diagnose von CML erforderlich sein können.
Deep Learning (DL)
Deep Learning, ein Teilgebiet der KI und des maschinellen Lernens, widmet sich dem Training neuronaler Netze, um aus umfangreichen Datensätzen zu lernen und Vorhersagen oder Entscheidungen zu treffen. Dieser fortschrittliche Ansatz ermöglicht die Erkennung von Mustern und präzise Vorhersagen in Bereichen wie der Bild- und Spracherkennung sowie der Verarbeitung natürlicher Sprache.
Maschinelles Lernen (ML) ist ein Teilbereich der künstlichen Intelligenz (KI), der sich auf die Entwicklung von Algorithmen und Modellen konzentriert, die es Computern ermöglichen, ohne explizite Programmierung zu lernen und ihre Leistung bei Aufgaben zu verbessern. Maschinelle Lernsysteme sammeln Informationen aus Daten und verbessern ihre Leistung schrittweise durch Erfahrung. Neuronale Netze sind eine Art von maschineller Lerntechnologie, die die Struktur und Funktionsweise der Netzwerke in den biologischen neuronalen Netzen des menschlichen Gehirns nachahmt. Sie bestehen aus verknüpften Knoten, die Informationen analysieren und weiterleiten, was ihnen ermöglicht, aus Datensätzen zu lernen.

Kosten- und Verlustfunktion
Ziel der DL-Modellentwicklung ist die Verringerung des Fehlers zwischen Vorhersagen und tatsächlichen Werten. Dies wird durch die Verwendung von Verlustfunktionen erreicht, die mit jedem Trainingsbeispiel verknüpft sind. Der Durchschnitt der Werte der Verlustfunktion über alle Datenproben ist die Kostenfunktion. Die Kostenfunktion wird optimiert, um den DL-Fehler zu minimieren. Durch die Optimierung der Kostenfunktion können wir die besten Ergebnisse im DL erzielen.
Metrik
Die Wirksamkeit von Algorithmen zur Interpretation medizinischer Bilder wird mit einer Vielzahl von Metriken bewertet. Die Tabelle, die zur Visualisierung der Algorithmenleistung und zur Bestimmung mehrerer Bewertungsmetriken verwendet wird, wird als Verwirrungsmatrix bezeichnet. Verwirrungsmatrizen werden verwendet, um DL-Modelle zu bewerten und eine realistischere Ansicht ihrer Leistung zu geben. Das Ergebnis könnte zwei oder mehr Klassen haben. In der Tabelle gibt es vier mögliche Kombinationen von vorhergesagtem und tatsächlichem Wert.
Mittlere durchschnittliche Präzision
Mean Average Precision (mAP) ist eine Metrik zur Bewertung von Objekterkennungsmodellen. Verwirrungsmatrix, Intersection over Union (IoU), Rückruf und Präzision sind die Submetriken, die das Rückgrat der Formel für die mAP-Genauigkeit bilden. Die mAP wird berechnet, indem die durchschnittliche Präzision (AP) für jede Klasse gefunden und dann über eine Anzahl von Klassen gemittelt wird.

Der HS Analysis Touch
Eine Schlüsseltechnologie der automatischen Interpretation von Gewebeproben in der HS Analysis-Software ist die neueste künstliche Intelligenz. Deep Learning bewertet ein statistisches Vorhersagemodell für die CML-Analyse, indem Algorithmen verwendet werden, um „zu lernen“, wie die CML-Erkrankung erkannt wird, und Zugang zu Variablen zu erhalten, die die CML-Überlebensrate vorhersagen. Einige Beispiele umfassen Quantifizierung, Durchmesserberechnungen und die Trennung gesunder und ungesunder Zellen.
Die HS Analysis-Software kann medizinischen Fachkräften dabei helfen, Störungen zu diagnostizieren, indem hochauflösende Bilder von Geweben und Organen untersucht werden. Und da das Programm Daten über den aktuellen Zustand des Patienten generiert, wird es den Ärzten helfen, geeignete Therapien auszuwählen. Einige Beispiele für Behandlungen werden später in diesem Blog besprochen.
Die folgenden Bilder zeigen die Benutzeroberfläche des HS Analysis KIT, in der der Projektname, die Werkzeugleiste und Registerkarten usw. zu sehen sind.
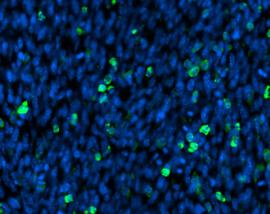
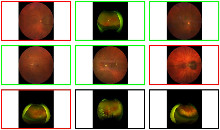
Innerhalb der Interessensbereiche werden Zeichnungen mit manuellen Benutzerannotationen erstellt. Nach dem Studium der Annotationen wendet das Deep-Learning-Modell diese auf mehrere größere Interessensbereiche an. Die einzelne Zelle wird entsprechend ihrem Typ annotiert. Zum Beispiel zeigen die folgenden Bilder, dass die Blutzellen in Gelb zu sehen sind, die gesunden Zellen durch Rot und die ungesunden durch Grün unterschieden werden. Ihre Klassen werden später erklärt.
Versteckte \ Sichtbare Blutzellen


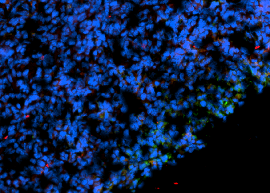
Neutrophiler Extrazellulärer Fallen „NET“ (Rosafarbene Struktur)


Neutrophile Extrazelluläre Fallen (NETs) sind spezialisierte Zellen des Immunsystems, insbesondere neutrophile Granulozyten, die eine entscheidende Rolle bei der Immunabwehr spielen. NETs sind Netzwerke von DNA-Fäden, die mit antimikrobiellen Proteinen und zellulären Komponenten gefüllt sind. Sie werden von neutrophilen Granulozyten freigesetzt, um Krankheitserreger zu erfassen und zu bekämpfen.
Bei der Aktivierung von neutrophilen Granulozyten, beispielsweise als Reaktion auf eine Infektion oder Entzündung, werden NETs freigesetzt. Die neutrophilen Granulozyten wickeln ihre chromosomale DNA ab und extrudieren ihr chromosomales DNA-Material, wodurch antimikrobielle Proteine freigesetzt werden, die Bakterien, Pilze und andere Krankheitserreger abtöten können. Die NETs bilden ein Netzwerk, das die Krankheitserreger einfängt und deren weitere Ausbreitung verhindert.
Während NETs eine wichtige Funktion bei der Abwehr von Infektionen haben, können sie auch zu bestimmten immunvermittelten Erkrankungen beitragen. Wenn NETs übermäßig aktiviert werden oder nicht effizient abgebaut werden, können sie zu einer übertriebenen Entzündungsreaktion und Schädigung des umliegenden Gewebes führen. NETs wurden mit verschiedenen Erkrankungen in Verbindung gebracht, darunter Autoimmunerkrankungen, Gefäßerkrankungen und andere entzündliche Zustände.
Die Forschung zu NETs und ihrer Rolle in der Immunabwehr und bei Krankheitsprozessen ist ein aktives Gebiet der medizinischen Forschung. Eine gezielte Modulation von NETs könnte potenziell neue Ansätze zur Behandlung von Infektionen und immunvermittelten Störungen bieten.
Pseudo-Gaucher-Zellen (Lila Struktur)


Pseudo-Gaucher-Zellen, auch bekannt als Pseudo-Gaucher-Makrophagen oder Pseudo-Gaucher-Zellen, sind eine Art von Zellen, die bei bestimmten Erkrankungen beobachtet werden und dem Erscheinungsbild der bei Morbus Gaucher beobachteten Zellen ähneln. Im Gegensatz zu echten Gaucher-Zellen repräsentieren Pseudo-Gaucher-Zellen jedoch nicht die Ansammlung von Glukozerebrosid, das für Morbus Gaucher charakteristisch ist.
Pseudo-Gaucher-Zellen sind typischerweise Makrophagen, die lipidgefüllte Vakuolen enthalten, wodurch sie ein ähnliches „knitteriges Seidenpapier“ oder „zerknittertes Seide“-Aussehen haben wie bei Gaucher-Zellen. Diese Zellen können durch mikroskopische Untersuchung von betroffenen Geweben oder Flüssigkeitsproben identifiziert werden.
Pseudo-Gaucher-Zellen können bei verschiedenen Erkrankungen gefunden werden, einschließlich lysosomaler Speicherkrankheiten, die nicht Morbus Gaucher sind, wie Morbus Niemann-Pick, Morbus Tay-Sachs und andere Lipidspeicherkrankheiten. Darüber hinaus können sie bei bestimmten nicht-lysosomalen Speicherkrankheiten und entzündlichen Zuständen beobachtet werden.
Das Vorhandensein von Pseudo-Gaucher-Zellen kann im diagnostischen Prozess hilfreich sein, da ihre Identifizierung auf die Notwendigkeit weiterer Untersuchungen hinweisen kann, um zwischen verschiedenen Krankheiten zu unterscheiden. Es ist jedoch wichtig zu beachten, dass das Vorhandensein von Pseudo-Gaucher-Zellen allein nicht diagnostisch für eine bestimmte Störung ist und weitere Tests normalerweise erforderlich sind, um eine genaue Diagnose zu stellen.
Megakaryozyt (Dunkelgrüne Struktur)

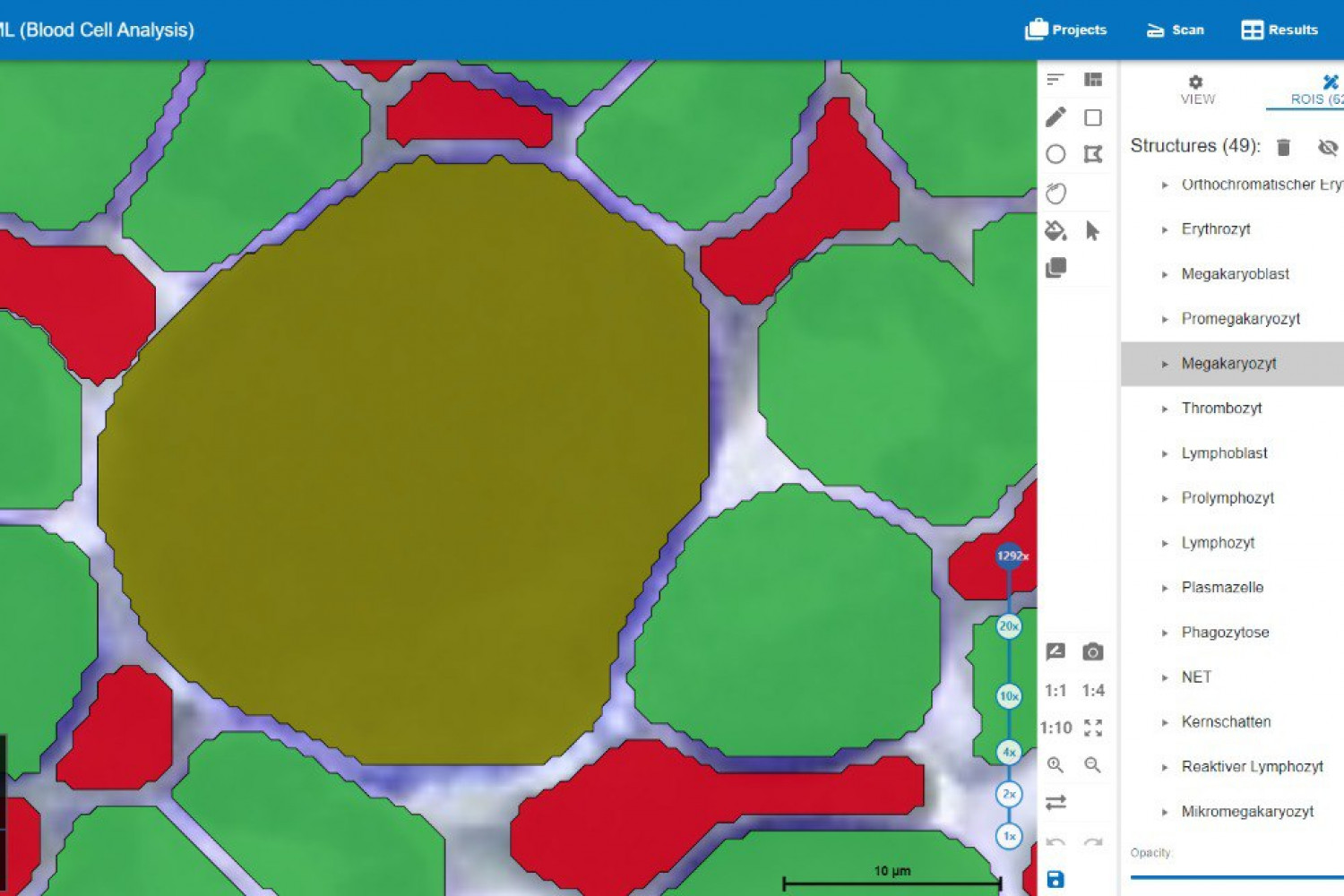
Ein Megakaryozyt ist eine große Knochenmarkszelle, die für die Produktion von Blutplättchen verantwortlich ist, die für die Blutgerinnung unerlässlich sind. Megakaryozyten sind Teil der hämatopoetischen Zelllinie, insbesondere der myeloischen Linie.
Megakaryozyten sind in ihrem Erscheinungsbild und ihrer Struktur einzigartig. Sie sind große, mehrkernige Zellen, was bedeutet, dass sie mehrere Kerne innerhalb eines einzigen Zellkörpers haben. Die Kerne der Megakaryozyten durchlaufen die Endomitose, einen Prozess, bei dem die DNA-Replikation ohne Zellteilung stattfindet, was zu einer polyploiden Zelle mit mehreren Chromosomensätzen führt.
Im Knochenmark befinden sich Megakaryozyten in speziellen Bereichen, den sogenannten Megakaryozyteninseln oder -nischen. Diese Nischen bieten eine Umgebung, die die Blutplättchenproduktion fördert. Megakaryozyten erstrecken lange, verzweigte Zytoplasmaausstülpungen, sogenannte Proplättchen, in Blutgefäße, wo sie fragmentieren und Blutplättchen bilden. Jeder Megakaryozyt kann zahlreiche Blutplättchen produzieren, die in den Blutkreislauf freigesetzt werden, um ihre wichtige Rolle bei der Blutgerinnung zu erfüllen.
Megakaryozyten werden durch verschiedene Wachstumsfaktoren und Hormone wie Thrombopoietin reguliert, das ihre Entwicklung und Reifung stimuliert. Abnormalitäten in der Produktion oder Funktion von Megakaryozyten können zu Blutplättchenstörungen wie Thrombozytopenie (niedrige Blutplättchenzahl) oder Thrombozytose (hohe Blutplättchenzahl) führen.
Das Studium der Megakaryozyten und ihrer Rolle bei der Blutplättchenproduktion ist wichtig, um Störungen der Blutgerinnung zu verstehen und Behandlungen für verwandte Erkrankungen zu entwickeln.
Darüber hinaus können wir in Bezug auf die Klassen die vielen verschiedenen Klassifikationen der von der HS Analysis-Software erkannten ungesunden Zellen sehen. Wie bei den anderen Annotationen müssen auch diese Klassen zunächst annotiert werden, bevor sie mit dem Deep-Learning-Modell trainiert werden. Es gibt über 40 CML-Klassen, aus denen das HS Analysis KIT lernen kann. Diese unterscheiden sich durch ihre Namen und Farben der annotierten Struktur. Zum Beispiel reichen sie vom jungen/alten Megakaryozyt bis hin zum sehr häufigen und dünnen Spinnennetzartigen NET.
Die Hauptbehandlung für chronische myeloische Leukämie ist ein Tyrosinkinase-Inhibitor (TKI). Forschungseinrichtungen und Pharmaunternehmen können testen, ob TKIs die Bildung von NETs, die von CML-Patienten gewonnen wurden, fördern oder verringern.
Die Anzahl der reifen/unreifen Megakaryozyten, die als Blasten bekannt sind, kann bestimmen, in welcher der 3 Phasen sich der CML-Patient derzeit befindet.


In Bezug auf Laborergebnisse, nachdem das Modell trainiert wurde und das gesamte Projekt durchlaufen ist, liefert uns die HS Analysis-Software die Informationen, die wir benötigen, um den Schweregrad und den Zustand in einem Tabellenformat zu verstehen, wie z. B. den Namen der Dateien, Anzahl der Objekte (Strukturen) wie gesunde und ungesunde Zellen, die Klassen der ungesunden Zellen, die Abmessungen der analysierten Bereiche wie Quadratmeter und Durchmesser der Zellgrößen und deren Durchschnitt usw.
Oder wir können die Deep-Learning-Analyse einer einzelnen Struktur wie gesunde Zellen oder einer/mehrerer ihrer Unterstrukturen wie Neutrophile Extrazelluläre Fallen (NETs) spezifizieren.
Durch die Nutzung der proprietären Deep-Learning-Software von HS Analysis und ihrer Laborauswertung können Ärzte beruhigt sein, da diese genauen Ergebnisse bei der Entscheidungsfindung für Patienten mit chronischer myeloischer Leukämie oder einem Forschungszentrum, das nach besseren Einblicken in die chronische myeloische Leukämie sucht, helfen können.
Der Bericht über die Knochenmarkmorphologie CMLpead wird als digitaler Bericht perfekt in die Software HSA KIT integriert. Die Zahlen aus der Quantifizierung mit HSA KIT werden in den Bericht geschrieben und bilden die Grundlage für evidenzbasierte Medizin. Auf der Grundlage dieses Berichts erhalten die Ärzte einen KI-Assistenten und Vorschläge zur möglichen Entwicklung und zum Status des Patienten.


CML-Patienten können beruhigt sein, da die Zusammenarbeit der Ärzte und medizinischen Forscher am Universitätsklinikum Erlangen und der Spezialisten für Bildanalyse und künstliche Intelligenz in der digitalen Pathologie bei HS Analysis den Weg zur Entdeckung besserer Medikamente deutlich voranbringen wird und nicht zu vergessen die Reduzierung der Sterblichkeitsrate bei chronischer myeloischer Leukämie.
Auswahl des Datensatzes
Nach Erstellung von GTD wurden die folgenden Tabellen beim Training des 3-Klassen-KI-Modells und des erythrozytären und leukozytären KI-Modells verwendet:
| Modelltyp | Epochen | Lernrate | Batch-Größe | Kachelgröße |
| Instanzsegmentierung | 100 | 0,0001 | 2 | 512 |
Die Einstellungen für das 3-Klassen-KI-Training
| Modelltyp | Epochen | Lernrate | Batch-Größe | Kachelgröße |
| Instanzsegmentierung | 50 | 0,0001 | 1 | 256 |
Die Einstellungen für das erythrozytäre und leukozytäre KI-Training
Framework
Das Deep-Learning-Modellierungs-Framework PyTorch hat sich als sehr beliebt und effektiv erwiesen. Diese auf Torch basierende Open-Source-Maschinenlern-Bibliothek wurde entwickelt, um die Implementierung tiefer neuronaler Netzwerke zu beschleunigen und flexibler zu gestalten. PyTorch ist eine auf Torch und Python basierende Deep-Learning-Tensor-Bibliothek, die hauptsächlich in CPU- und GPU-Anwendungen eingesetzt wird. Torch ist eine Open-Source-ML-Bibliothek zur Erstellung tiefer neuronaler Netzwerke und ist in der Skriptsprache Lua geschrieben.
Erstellung von Ground-Truth-Daten
Um ein Modell im Deep Learning zu trainieren, wird GTD benötigt, das durch einfaches Erstellen von Basis-ROIs und Annotieren der vorhandenen Zellen innerhalb der Basis-ROIs erreicht wird. Die Anzahl der Zellen im verfügbaren GTD-Formular (siehe Tab.1) betrug 10 Dateien zusammen mit + 800.000 GTD unter Verwendung der proprietären Software HSA KIT. Es gibt verschiedene Phasen bei der Erstellung von GTD. Die Carl Zeiss Image Data File enthält eine WSI-Datei, die zunächst in das HSA KIT geladen wird (CZI). Die Annotationen werden innerhalb des ROI in Bezug auf eine Funktion des HSA KIT festgelegt. Anschließend wird die Blutstruktur annotiert, und diese Struktur hat zwei Unterstrukturen, nämlich Erythrozyten und Leukozyten, wobei die Leukozyten weiter in 45 verschiedene Klassen unterteilt sind, darunter (NET, Pseudo-Gaucher-Zelle und Megakaryozyt), und die Anzahl der Klassen beträgt (5399, 97 und 149) bzw. Die Anzahl und Qualität dieser Annotationen hängt von der Lage, Klarheit und Größe des Basis-ROIs ab. Unten finden Sie eine Aufschlüsselung der Klassenverteilung zusammen mit den entsprechenden Prozentsätzen.
| Klassen | Leukozyten | Erythrozyten | Megakaryozyt | NET | Pseudo-Gaucher-Zelle | Segmentkernige Granulozyten | Stabkernige Granulozyten |
| Alle Daten | 274.553 | 617.709 | 149 | 5.399 | 97 | 4194 | 1678 |
| Verwendete Daten | 48.887 | 88.947 | 59 | 4.587 | 93 | 4194 | 1678 |
Die verfügbaren und verwendeten Datenmengen
Fortschrittlichste Instanzsegmentierung für CML weltweit
Die Software HSA KIT umfasst HyperCMLNet, die weltweit fortschrittlichste Instanzsegmentierung für CML. HyperCMLNet als Deep-Learning-Technik wurde bei der HS Analysis GmbH entwickelt und basiert auf Mask R-CNN und Vision Transformer (ViT).
In der Software HSA KIT erstellen und verwalten Datenwissenschaftler nicht nur hochwertige Ground-Truth-Daten oder trainieren und verwalten entsprechende Deep-Learning-Modelle, sondern sie können die Modelle auch mit Metriken und Overlays vergleichen.
Unten sehen Sie die Metriken des ersten HyperCMLNet (Typ 1) basierend auf Mask R-CNN sowie des zweiten HyperCMLNet (Typ 2) basierend auf Vision Transformer (ViT). Das HyperCMLNet trennt im ersten Schritt Erythrozyten von Leukozyten und im zweiten Schritt alle Klassen von Leukozyten. Die Ärzte können vollautomatisch alle Klassen von CML, z.B. NET, Pseudo-Gaucher-Zelle und Megakaryozyt, etc. sowie Erythrozyten erkennen. Die Klassen wurden auf 137.834 GTD trainiert. Diese Modelle wurden in der Software HSA KIT sowohl mit den Metriken mAP als auch Verlust bewertet.
Interpretation und Validierung des Ergebnisses
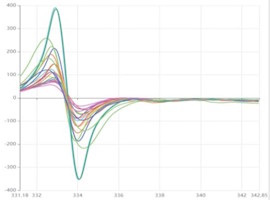
Die folgenden Daten zeigen den Vergleich sowohl der Verlust- als auch der mAP-Bewertungen der Ergebnisse der Instanzsegmentierung, die von beiden trainierten (Typ 1 & Typ 2) der HyperCMLNet-Architekturen für Erythrozyten und Leukozyten und die 3 Klassifikationen durchgeführt wurden. Die folgende Tabelle zeigt die tatsächlich erzielten Ergebnisse des Modelltrainings.
Im Falle von Verlust, einer Funktion, die verringert werden muss, um ein ideales Deep-Learning-Modell zu erhalten. Der Verlustwert würde eine große Zahl produzieren, wenn seine Vorhersagen zu stark von den tatsächlichen Ergebnissen abweichen. Die Grafik zeigt, dass das auf Mask R-CNN (Typ 1) basierende HyperCMLNet einen niedrigeren Verlust aufweist, verglichen mit dem auf ViT basierenden HyperCMLNet (Typ 2). Wir können sehen, dass das auf Mask-R-CNN basierende HyperCMLNet (Typ 1) die Verlustmetriken von 3 % und 11 % aufweist, verglichen mit den höheren Verlustmetriken des auf ViT basierenden HyperCMLNet (Typ 2), die 4 % und 13 % betragen. In Bezug auf Verlust hat das HyperCMLNet (Typ 1) also die besten Verlustbewertungen von beiden Typen.
Im Fall von mAP, der Genauigkeit der Instanzsegmentierungserkennung, wird die Genauigkeit der Ergebnisse der Instanzsegmentierung unter Verwendung der mittleren durchschnittlichen Präzision (mAP) gemessen, einer weiteren Bewertungsmetrik. Je höher der mAP-Wert, desto genauer sind die Ergebnisse der Instanzsegmentierung. Die Grafik zeigt auch, dass diesmal das auf ViT basierende HyperCMLNet (Typ 2) bessere mAP-Werte von 94 % und 96 % aufweist, verglichen mit den niedrigeren mAP-Werten, die vom auf Mask R-CNN basierenden HyperCMLNet (Typ 1) erzielt wurden, die 86 % und 89 % betragen. In Bezug auf mAP hat das HyperCMLNet (Typ 2) also die besten mAP-Bewertungen von beiden Typen.
Visuelle Interpretation der Ergebnisse:
Nach dem AI-Training für ein beliebiges Modell endet es immer damit, dass der Benutzer mathematische Ergebnisse wie Grafiken oder numerische Ergebnisse erhält. Obwohl diese wichtig sind, wird eine weitere Bestimmung der Qualität des KI-Modells durch manuelle Überprüfung der Erkennung vorgenommen. Dies führt uns zu diesem Abschnitt der visuellen Interpretation der Ergebnisse, und dieser Abschnitt veranschaulicht die Visualisierung der Instanzsegmentierung beider (Typ 1 & Typ 2) KI-Modelle der CML-Bilder, die folgende Abbildung zeigt mehrere Bilder, die das Original und die (Vorhersagen) zeigen, die von den Netzwerken ermittelt wurden.
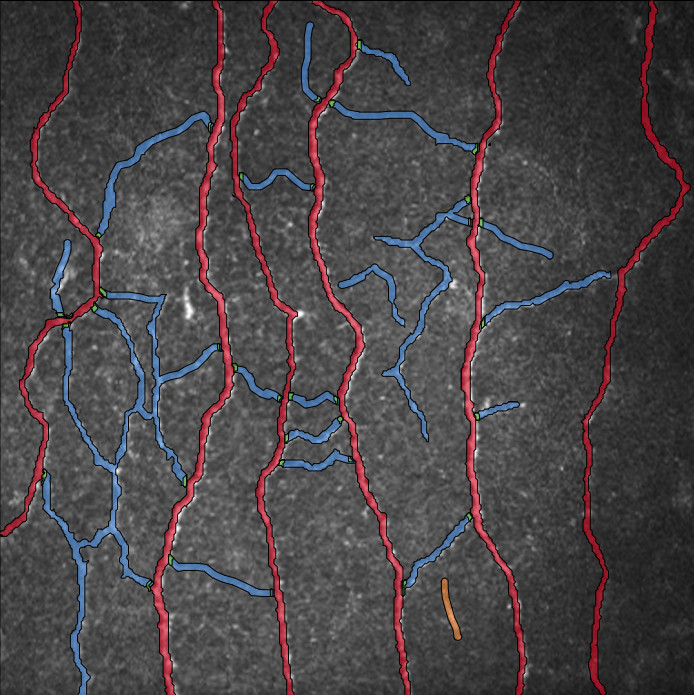
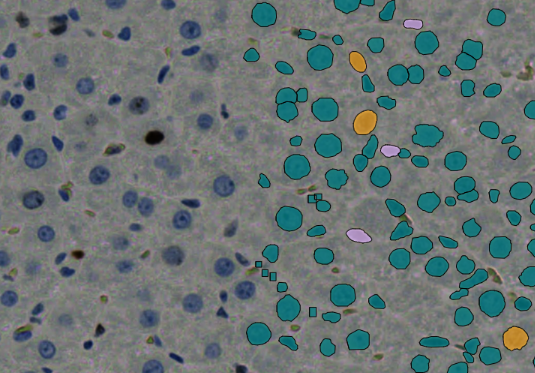
Die in der Begleitabbildung gezeigten Zellenerkennungsergebnisse, die aus Modelltrainings gewonnen wurden ((A) Erythrozyten und Leukozyten, (B) NET, (C) Megakaryozyt und (D) Pseudo), zeigen, dass beide (Typen 1 & Typ 2) von HyperCMLNet sehr ähnliche Erkennung aufweisen. Erstens zeigt, wie der Name schon sagt, die Spalte des Originalbilds den untrainierten, rohen und zellerkennungsfreien Teil des CML-Bildes. Das trainierte HyperCMLNet-Modell wurde auf einige der in der zweiten Spalte gezeigten Basis-ROIs angewendet (Typ 1). Diese Basis-ROIs zeigen die Erkennung von Erythrozyten und Leukozyten sowie die Unterstrukturen von Leukozyten.
Drittens wird das trainierte HyperCMLNet-Modell (Typ 2) in der dritten und letzten Spalte angezeigt und auf dieselben Basis-ROIs wie das (Typ 1) Modell angewendet. Beim Vergleich der Typen 1 und 2 in Bezug auf Erythrozyten und Leukozyten weisen beide Modelle eine hervorragende Erkennung von Erythrozyten und Leukozyten auf und besitzen ein hohes Maß an Detailtreue und Erkennungsgrenzen.
Die Brillanz der HyperCMLNet-Architektur war in der Lage, den Fehleranfälligen bei der Erkennung der Trennung einiger zusammengeklumpter Erythrozytenzellen sowie Leukozyten zu unterscheiden, obwohl deren Fehlerwahrscheinlichkeit geringer ist, da sie eine deutlich kleinere Fläche haben, insbesondere im Vergleich zu Erythrozyten. Dieses Projekt war sowohl groß als auch komplex. Um eng beieinander liegende Erythrozytenzellen zu unterscheiden, konnte das HyperCMLNet seine Ergebnisse klar und verständlich darstellen.
Auch hier ist die Qualität in beiden (Typ 1 & Typ 2) der HyperCMLNet-Designs bemerkenswert, wenn man die (Typ 1 & Typ 2) Modelle in Bezug auf NET-Erkennung vergleicht. Es hat die Fähigkeit, die Erkennung von Nicht-NET-Zellen zu ignorieren und sich nur auf die Erkennung von NET-Zellen zu konzentrieren.
Die Typen 1 und 2 werden mit Megakaryozyten und Pseudo verglichen. Beide (Typ 1 & Typ 2) scheinen Megakaryozyten und Pseudo mit hoher Präzision und Genauigkeit in allen Dateien zu erkennen und liefern eine hervorragende Erkennung. Dies liegt daran, dass beide (Typ 1 & Typ 2) große Datenmengen für das Training von HyperCMLNet (Typ 1 & Typ 2) verwendet haben. In fast allen Fällen zeigte das HyperCMLNet (Typ 2) jedoch etwas mehr Aufmerksamkeit für Details bei der Erkennung der CML-Strukturen im Vergleich zum HyperCMLNet (Typ 1). Die Abbildung unten zeigt die Visualisierung der Trainingsergebnisse beider Typen von HyperCMLNet.

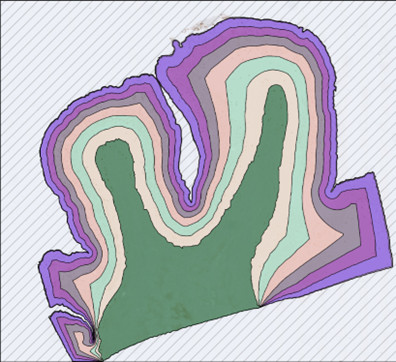
Fortschrittliches Heatmap-Tool von HSA KIT
Das erweiterte Heatmap-Tool zur Erkennung, das im HSA KIT verwendet wurde, wird in diesem Abschnitt verwendet, um die Ergebnisse der CML-Klassen miteinander zu vergleichen. Das fortschrittliche Heatmap-Tool im HSA KIT wird verwendet, um vorherzusagen, wo die Klassen der KI-Modelle am wahrscheinlichsten zu finden sind. Je wärmer (mehr rot) die Heatmap ist, desto höher ist die Wahrscheinlichkeit der Erkennung; umgekehrt, je kälter (mehr blau), desto unwahrscheinlicher wird die Erkennungsvorhersage auftreten.
Im Wesentlichen wird der Hotspot der von der KI generierten Annotationen mit dem ausgeklügelten Heatmap-Tool visualisiert. Es ist für KI-Visualisierungsprojekte wie das hier verwendete konzipiert, das die Ergebnisse mit vielen Details auf einer einzigen Folie anzeigt.
Die HyperCMLNet-ViT- (Typ 2) und HyperCMLNet-Mask-R-CNN- (Typ 1) Architekturen werden in der ersten Spalte und die Erythrozyten-, Leukozyten- und (B, C und D) Leukozytenklassen in der zweiten Spalte der folgenden Abbildung verwendet.
Wir können nicht wirklich sagen, welche der beiden Architekturen in (A) idealer ist, da es in Bezug auf die fortschrittliche Heatmap-Erkennung von Erythrozyten und Leukozyten-Mutterstrukturen kaum einen Unterschied zwischen ihnen gibt, aber beide Architekturen haben großartige Hotspot-Vorhersagen.
Im Fall von (B, C und D) war die Erkennungsqualität für beide Designs vergleichbar und lieferte ermutigende Ergebnisse, obwohl sie viele der gleichen Merkmale in Bezug auf präzise und genaue Heatmap-Erkennung aufwiesen.
Mit Hilfe des fortschrittlichen Heatmap-Tools im HSA KIT kann die Lokalisierung der Heatmap-Vorhersage durch Hinzufügen weiterer GTD weiter verbessert werden. Das fortschrittliche Heatmap-Tool bietet auch die Möglichkeit, den Intensitätsschieberegler so einzustellen, dass wir die Heatmap in verschiedenen Intervallen betrachten und die revolutionäre Erkennung der HyperCMLNet-Architektur vollständig schätzen können. Die folgende Abbildung zeigt die Visualisierung des erweiterten Heatmap-Tools von HSA KIT für die Trainingsergebnisse beider Typen von HyperCMLNet.

In dieser Abbildung wurden die Mutterstrukturen (Erythrozyten, Leukozyten) und die 3 Unterklassen der Leukozyten (NET, Pseudo-Gaucher-Zelle und Megakaryozyt) der chronischen myeloischen Leukämie mit Deep-Learning-Modellen erkannt und dann berechnet und validiert. Darüber hinaus wurde das Heatmap-Tool von HSA KIT verwendet, um diese Modelle zu visualisieren und zu bestimmen, wie dieselben Zellklassen mit unterschiedlicher Architektur erkannt wurden. HyperCMLNet (Typ 1), das auf der Mask-R-CNN-Architektur basiert, und HyperCMLNet (Typ 2), das auf der Vision Transformer-Architektur basiert, wurden beide auf GTD von CML implementiert. Das Ziel war es, die Erkennung mehrerer Klassen von CML auf beiden Architekturen in der Instanzsegmentierung im Deep Learning zu vergleichen.
Digitalisierung von histologischen Schnitten und Analyse mit HSA KIT
HSA importiert und arbeitet mit 3dhistech- und mirax-Dateien, die vom Hamamatsu-Scanner erstellt wurden. Sie passen perfekt zum HSA KIT.
Wenn Sie keinen Slide-Scanner haben und später einen erwerben möchten, können Sie die Slides manuell auf Ihrem Mikroskop digitalisieren und mit unserer günstigen und erschwinglichen Software (HSA SCAN M) manuelle WSI erstellen.

Die benutzerfreundliche Oberfläche der Software ermöglicht eine einfache Navigation und Anpassung der Scan-Einstellungen an spezifische Anforderungen. Darüber hinaus ermöglicht die Integration mit der HSA KIT-Software eine erweiterte Bildanalyse und Interpretation, die wertvolle Einblicke für Forschung und Diagnose liefert. Insgesamt bietet die HSA SCAN-Software eine umfassende Lösung für Labors, die ihren Slide-Digitalisierungsprozess rationalisieren und gleichzeitig die Qualitätskontrolle aufrechterhalten möchten.

HSA benötigt lediglich die Abmessungen des Mikroskops und alle Spezifikationen, und ein maßgeschneiderter Ständer und Motor für das Mikroskop werden produziert. Diese Technologie macht manuelle Anpassungen überflüssig und liefert konsistente Ergebnisse.
Um die Slides automatisch zu scannen, kann das vorhandene Mikroskop zu einer automatisierten Mikroskopstation (HSA SCAN A) aufgerüstet werden, die in kurzer Zeit eine kostengünstige, qualitativ hochwertige Leistung bietet.
Dies verwandelt Ihr Mikroskop in einen automatischen Scanner für eine kostengünstige, qualitativ hochwertige Leistung in kurzer Zeit.
HSA SCAN M

In diesem Video sehen Sie manuelles Scannen, das einen Slide mit der HSA SCAN-Software in eine Datei umwandelt, sodass es trainiert und dann automatisch von einem trainierten KI-Modell unter Verwendung der HSA KIT-Software erkannt werden kann.
Dies wird unterstützen bei:
- Genaues und schnelles Scannen
- Budget im Griff behalten: Weniger teuer als automatische Scanner
- Zeitsparend: Automatisiert Aufgaben, die manuelle Eingriffe erfordern
- Besserer Arbeitsablauf und qualitativ hochwertige Ergebnisse: Geringeres Risiko von Fehlern oder Inkonsistenzen
- Intuitive, benutzerfreundliche Steuerung: Jeder mit begrenzter Erfahrung mit Mikroskopen kann sie bedienen
HSA SCAN A

Im obigen Video sehen Sie ein Beispiel für einen automatischen Scanner (HSA SCAN A). Zögern Sie nicht, uns für weitere Informationen oder Bestellungen zu kontaktieren.
Dies wird unterstützen bei:
- Genaues und schnelles Scannen
- Budget im Griff behalten: Weniger teuer als automatische Scanner
- Zeitsparend: Automatisiert Aufgaben, die manuelle Eingriffe erfordern
- Besserer Arbeitsablauf und qualitativ hochwertige Ergebnisse: Geringeres Risiko von Fehlern oder Inkonsistenzen
- Intuitive, benutzerfreundliche Steuerung: Jeder mit begrenzter Erfahrung mit Mikroskopen kann sie bedienen