By leveraging the power of advanced technologies like big data and artificial intelligence (AI), healthcare providers can not only improve diagnosis accuracy but also enable a proactive approach that allows for early intervention and personalized preventive measures, ultimately leading to better overall health management. Integration of diverse sources of medical data for example, patient feedback, genetic data, and electronic health records can provide a holistic understanding of each patient’s unique circumstances and also lead to the discovery of new patterns and correlations that may further enhance medical research and advancements in biotechnology.
Multi-modal medical data fusion
In multi-modal technology, a single mode of the medical image can supplement the weakness of another mode to accurately evaluate the medical condition and obtain diagnostic information through the fusion of information from multiple modes. Multi-modal fusion is related to representations, and a process that focuses on using some architecture to merge representations of different single models is classified as fusion.

HSA Solutions to Medical Imaging
Medical imaging is an effective way for disease screening and lesion localisation, but mass data means the professional have to spend alot of time to analyze and draw conclusions that are mostly subjective. Use of Artificial Intelligence can not only speed up the procedure, but can reduce the chances of misdiagnosis. However there are some issues:
- Uneven Image quality: Some images are relatively poor in quality, with a lot of noise, blurred edges, and the background of the images is complex and inconsistent, thus it is very difficult to extract features and analyze them
- Lack of Standardisation: Large sets of data requires manual annotation by a professional, privacy protection of the patient, scarce and non-generalised data labelling
- Image inaccuracy: Medical images have multi-modal image channels with complex noise in contrast to conventional natural image segmentation. Real-time performance and higher image accuracy are necessary




HS Analysis uses Deep Learning algorithms that are well suited to take advantage of the complex and heterogeneous data types obtained in modern clinical practice to learn the extremely complex relationship between features and labels, so as to assist doctors to make relevant analyses and predictions.
Digitalisation of slides
The HSA KIT is extremely versatile and pairs well with some of the best automatic slide scanners like the Leica GT 450. It was designed specifically to seamlessly integrate with such devices and enable quick and precise slide scanning. The HSA KIT also offers cutting-edge features like automated slide handling and adjustable scanning parameters, further enhancing its incorporation within digital pathology infrastructure. Users can use these features to personalize their scans and streamline the scanning process to suit their needs. Professionals in disciplines like pathology and research will find the HSA KIT to be an excellent choice because of its seamless integration with their pre-existing technology.
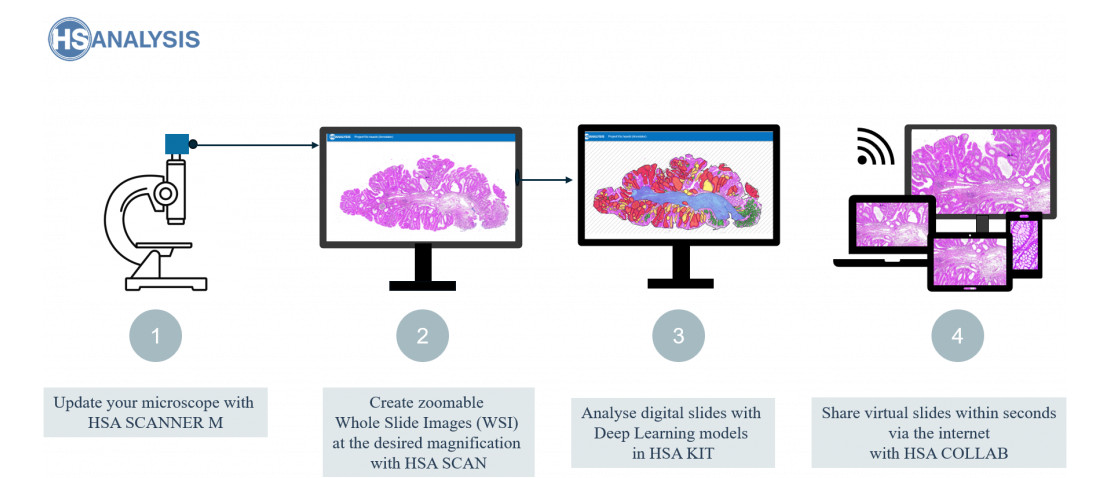
At the same time, HS Analysis provides an affordable alternative to other automatic scanners- HSA Scan M software. The slides can be manually digitalized directly on current microscope to create manual WSI (whole slide images). This allows for greater flexibility and control over the scanning process. Additionally, the HSA KIT can be used with a variety of microscope models, making it a versatile option for labs with multiple microscopes. The software is user-friendly and easy to navigate, allowing for efficient scanning and image processing.
Using Mass Spectrometry data
Metabolic reprogramming during the earliest stages of cancer growth initiates a radical change in cell chemistry. Evidence of oncogenesis can be found in subtle changes to the composition of tumors as they progress from abnormal cells to aggressive metastatic cancers. Cancer research has increasingly embraced mass spectrometry-based profiling of clinical specimens (such as tissues and biofluids). A frequent strategy is to describe differences in protein expression between tumor and healthy tissue or between blood from sick and healthy people.
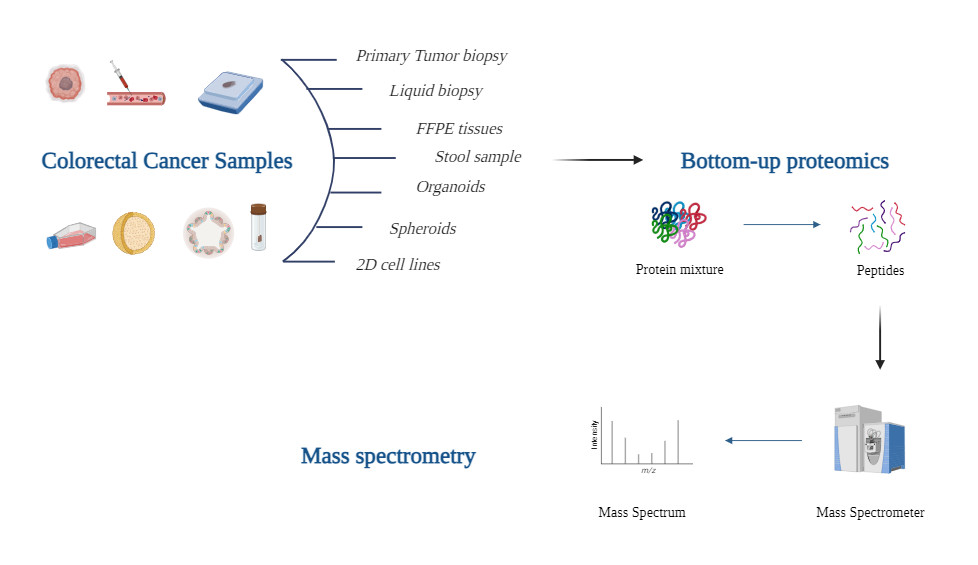
There are currently numerous forms of MSI techniques available, some prominent ones are:
- Liquid chromatography-mass spectrometry (LC-MS): utilized to obtain a global proteomic profile of a biological sample
- Laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS): ICP-MS is an analytical technique widely used for quantitative element determination in liquid samples. Laser ablation is used for solid sample introduction to ICP-MS
- Probe electrospray ionization-mass spectrometry (PESI-MS): rapidly visualize mass spectra of small, surgically obtained tissue sample
MS Analysis in HSA KIT
There are many different ways to visualize and understand MS data in HSA KIT. The program can currently handle .d (Bruker) and .mzML files seamlessly. Additionally, the data from results can be exported in various formats for further analysis or sharing with different collaborators. The flexibility of HSA KIT allows for seamless integration with other analytical tools and workflows. New file formats are being added to the program to further its functionality.
Moreover, the built-in statistical analysis tools can be used to identify trends and patterns in the data. The continuous updates and improvements to HSA KIT ensure that users have access to the latest features and capabilities in MS data analysis. It is possible to perform proteome analyses on tumor tissues and compare the outcomes with those of healthy tissues. This can provide valuable insights into the molecular mechanisms underlying cancer development and progression. Additionally, HSA KIT offers user-friendly interfaces and customizable settings to cater to individual research needs.
MS Viewer: for importation, visualization, and interpretation of MS data, where raw data is combined with FASTA files to provide detailed information about the proteins identified in the sample, and for analysis of protein-protein interactions to gain insight into biological pathways and networks.
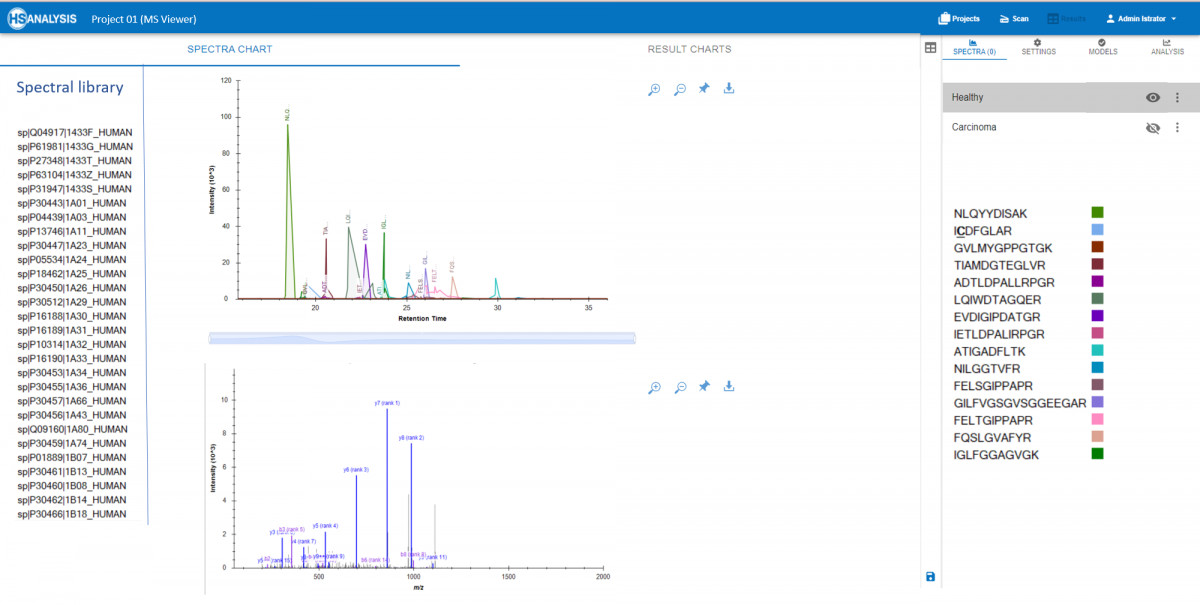
There is a possibility to view peaks and spectra of peptides in real time, alongwith corresponding proteins from the spectral library. Additionally, the MS Viewer module allows for the comparison of multiple samples to identify differences in protein expression levels or modifications. This comprehensive tool is essential for researchers looking to uncover key insights from their mass spectrometry experiments. One application is to compare MS peaks in Healthy and Cancerous tissues where any misbehavior of peptides can be easily spotted and analysed further.
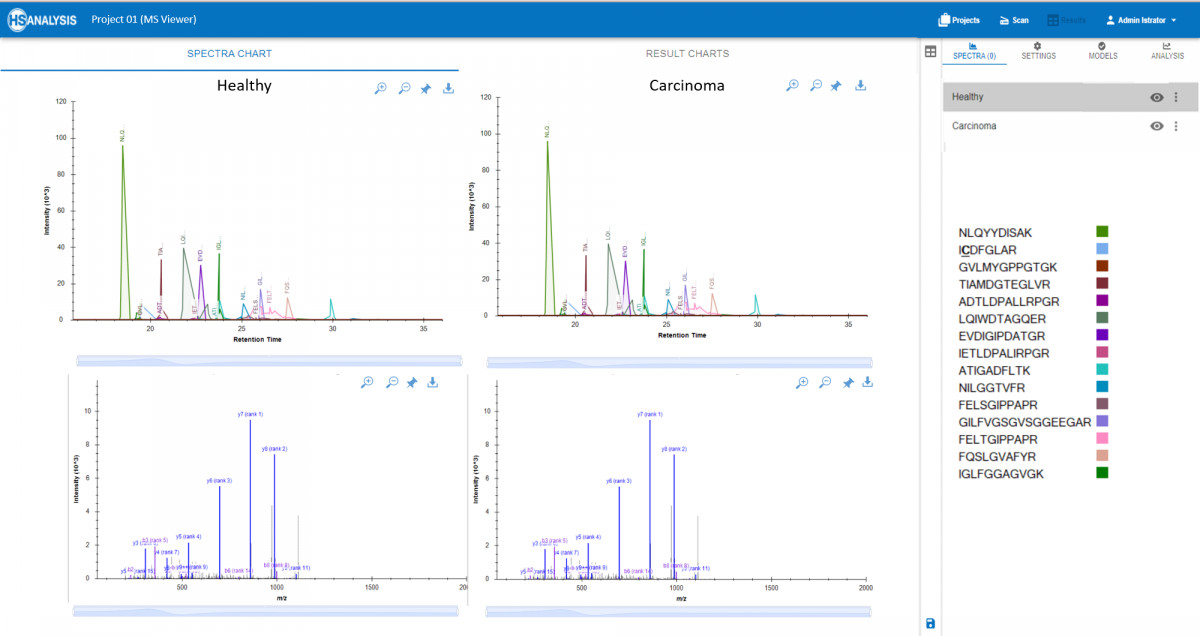
MS Viewer not only gives a visualisation experience, but also generates a report at the end which contains information about each peptide (precursor) present in a protein with it`s abundance and other relevant factors. Additionally, there is an opportunity to obtain a comparative quantification of data where significant differences observed in two files can be highlighted.
MS Analyser: Following the results obtained from MS Viewer, this module is a powerful tool for studying protein structures and functions. Main focus is on finding peptide positions and mapping them onto the whole protein sequence to better understand protein interactions within networks. Additionally, MS Analyser can also be used to determine post-translational modifications of proteins, providing further insight into their functions and regulatory mechanisms. By analyzing peptide positions and mapping them onto the protein sequence, researchers can uncover key residues involved in protein interactions and signaling pathways.
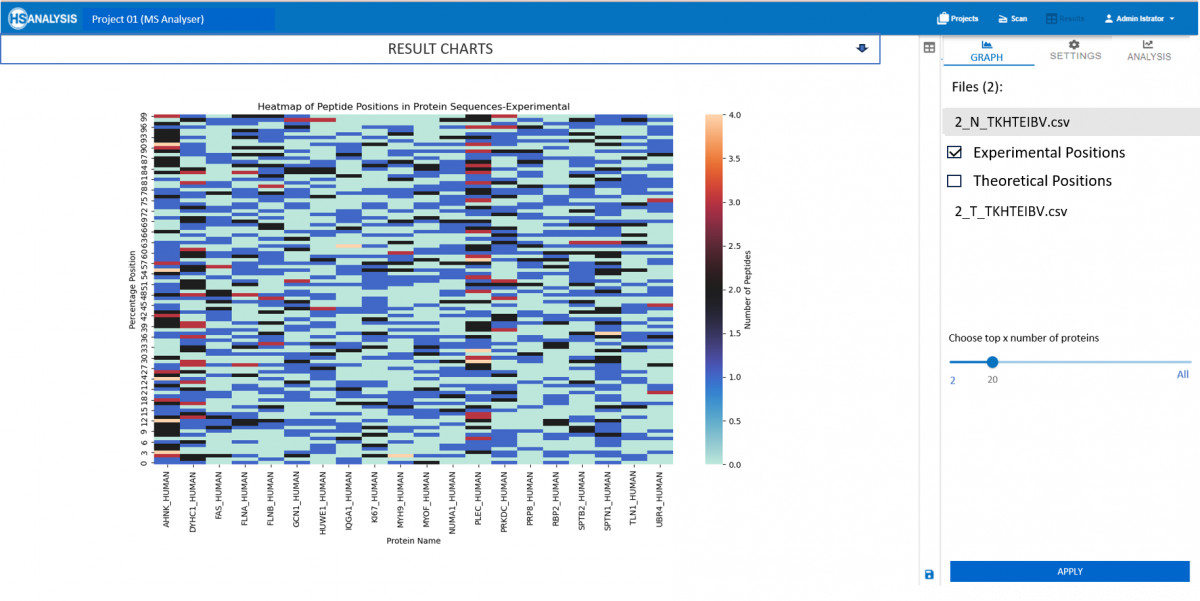
Experimental results correspond to the input .csv file which was produced as a result by our MS Viewer, It contains important information about quality and quantity of peptides. This data can then be further analyzed using bioinformatics tools to identify patterns and relationships between proteins. By integrating multiple sources of data, a comprehensive understanding of protein function and regulation within biological systems can be obtained.
As for the theoretical results, these are the expected behaviour of peptides once their respective protein is digested with trypsin. This provides valuable insights into expected protein structure and its behavior. Additionally, this data can be used to validate experimental results and guide future research directions in the field of proteomics.
Most viable use case of this module is to check which peptides are missing or in higher concentration that expected in experimental results.
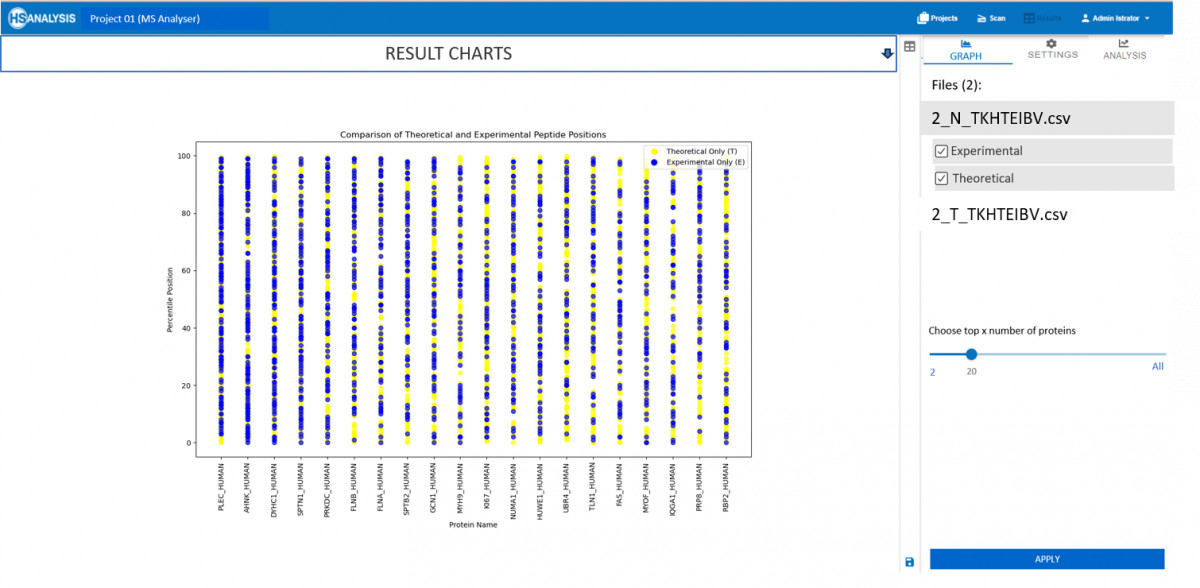
Now that the actual and expected peptide positions are obtained, a detailed overview can be found automatically in the analysis tab. Most importantly the comparison of this type of result can further be compared for two different files, for example, healthy vs cancerous tissues.
As a summary, MS Viewer is primarily a tool to study protein expression while MS Analyser deals with position of peptides. A comination of both these modules gives an end-to-end proteomics solution, especially for cancer studies where huge data needs to be interpreted in a simplest form.
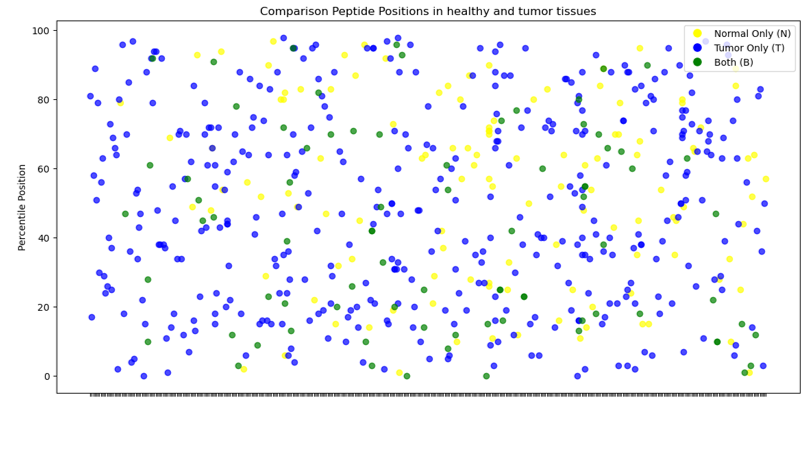
In summary, the MS Analyser model has shown a capability to differentiate between normal and tumor tissue at a molecular level, with variations in protein and peptide counts being noted. Some proteins manifest with higher peptide counts, although this may not be consistent across all cases of CRC. Lastly, graphical representations suggest certain peptides’ absence in tumor tissues compared to normal tissues, pointing towards specific molecular changes associated with cancerous transformations.
The abundance of peptides alone cannot reveal much information about their behavior unless it is studied with their intensity. Theoretically, the peptides belonging to the same protein must have the same stoichiometry i.e. they must be present in equal molar amounts. However, their detection in Mass Spectrometers is influenced by the presence of other precursor ions.

Research is a time-consuming process that necessitates long-term answers. The use of such libraries restricts the ability to integrate with future ambitions. Furthermore, relying only on pre-existing libraries may limit one’s ability to adapt to and incorporate new technologies or approaches that arise in the future. It is critical for researchers and developers to be able to experiment with new methodologies and tailor their solutions to changing research needs.
HSA KIT offers seamless integration with existing laboratory equipment and software systems, streamlining the workflow and maximizing efficiency. This allows researchers and developers to focus more on their scientific goals rather than technical challenges.
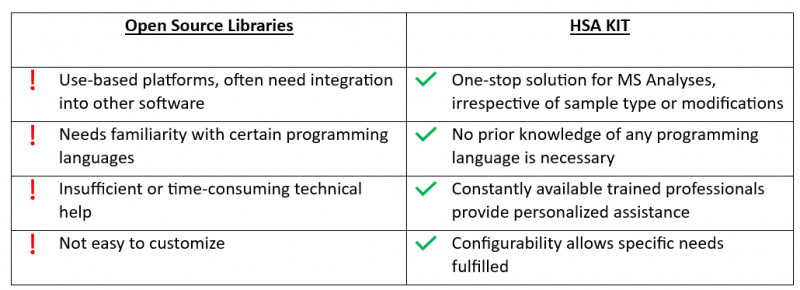
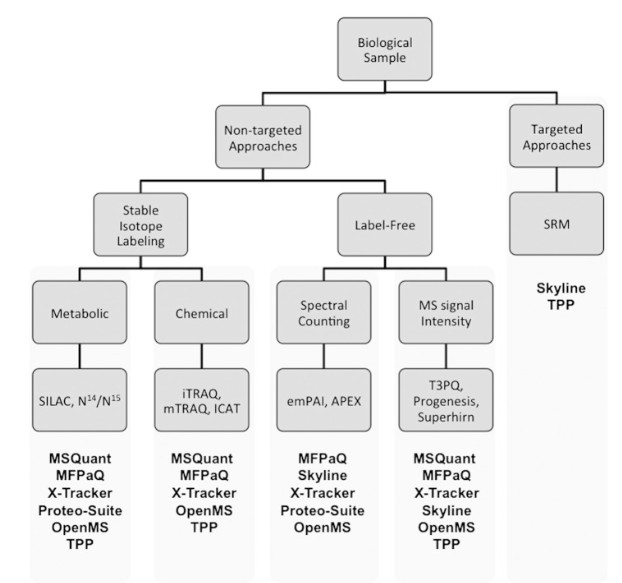
A community of bioinformaticians and software developers develop and maintain a wide range of software solutions covering majority elements of MS data analysis.
Common MS data processing tasks include theoretical proteome analysis, raw spectral processing, file format conversions, identification statistic generation, and storage/visualization of raw data, identification, and quantitation results.

Open source software has certain drawbacks, such as limited compatibility with certain data formats, a difficult user interface, and a lack of thorough technical support.
In addition to streamlining the analysis process for mass spectrometry, HSA KIT offers an adaptable platform for incorporating deep learning techniques. This extends its potential beyond conventional mass spectrometry applications by enabling the modification of modules to suit a variety of applications like illness detection, food toxicity certification, or principal component analysis. Not to mention the capability to work with various data types and, if needed, carry out automatic file conversions.
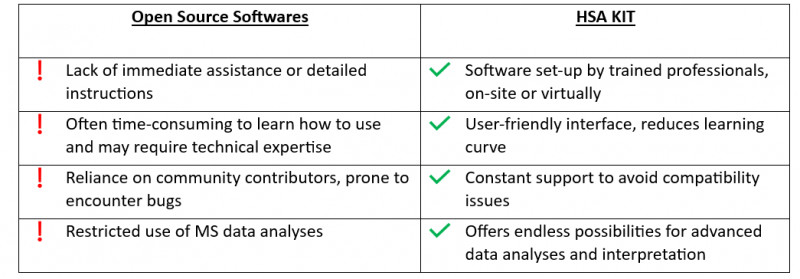
Medical Image fusion
Multi-modal medical image fusion based on deep learning can be used to effectively extract and integrate the feature information of different modes, improve the clinical applicability of medical images in the diagnosis and evaluation of medical problems, and provide quantitative analysis, real-time monitoring, and treatment planning for doctors and researchers.
There are currently two main approaches to multi-modal tasks: light fusion and heavy fusion.
- Light fusion: usually effortless, such as the vector inner product, as represented by CLIP and ALIGN, which use a two-tower structure focusing on multi-modal alignment to facilitate text matching, retrieval, and other downstream tasks
- Heavy fusion: based on pre-trained Transformers, as represented by OSCAR, UNITER, VINVL, etc. Heavy fusion can interpret VQA, captions, and other downstream tasks that require information fusion and understanding, which the ALIGN algorithm cannot perform
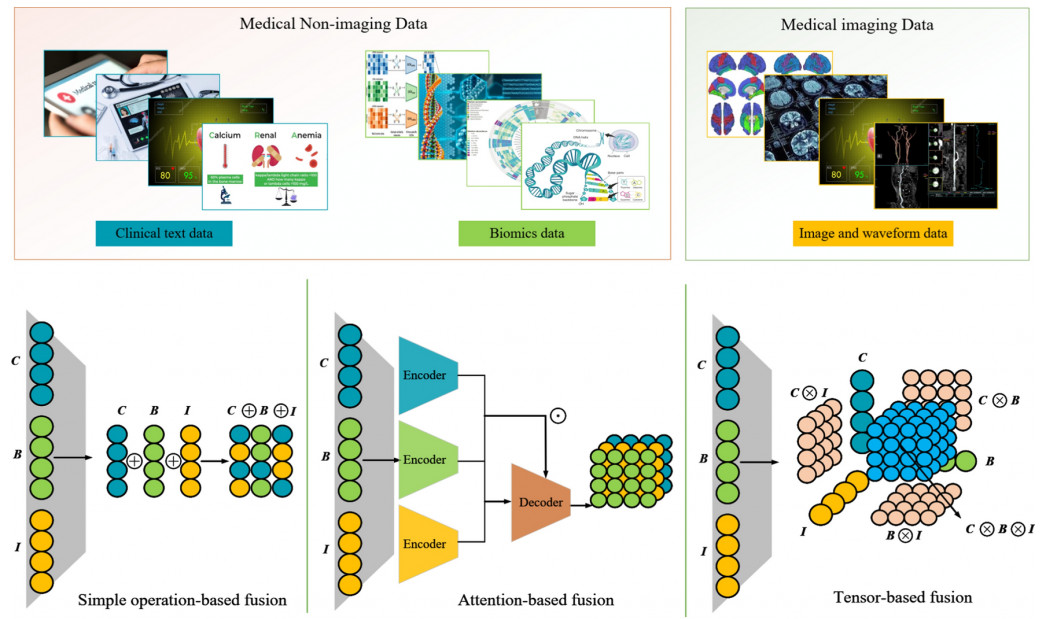
Fusion methods can be divided into late and early fusion according to their different locations.
- Early fusion: Its advantage is in taking advantage of early in the analysis correlations between several characteristics from several modalities. When there is a strong correlation between modalities, such as when merging audio and visual inputs for speech recognition, this method is recommended
- Late fusion: Each modality must have its own models trained, and the results must then be combined. After combining feature vectors from various independent sources, late fusion makes decisions based on the joint features that are produced
- Hybrid fusion: to optimize the advantages of early and late fusion. But it also adds to the model’s complexity and increases the difficulty of training. In situations when flexibility is required, hybrid fusion approaches might be a better option because of the adaptability of deep learning model structures
There are three methods for the fusion of text and image: simple operation-based, attention-based, and tensor-based approaches.
Hybrid Architecture of CNN/RNN
CNNs are a family of discriminative models that have shown efficacy in the majority of computer vision applications, including object detection, semantic segmentation, image classification, and so forth. They take advantage of the spatial features of visual input streams. RNN networks are more suited to reflect the temporal relationships of complicated tasks like speech recognition and machine translation because they can model sequential data and time series better than CNN can. To put it another way, RNN is better at considering correlations in spatiotemporal information, but CNN is more effective at extracting visual characteristics from static data like pictures, depth maps, etc. For a variety of applications, attempts have been made to combine CNN and RNN architectures. Even more specifically, RNN predicts the subsequent state in the observed event sequence based on previously hidden states, whereas CNN employs convolutional kernels to transform spatial data.
Multi-Modal Analyser (MMA) in HSA KIT
The fusion of two modalities in the CRC study, image analysis and proteomics sequence analysis, was conducted using a decision-level fusion methodology. Two distinct modules were trained and validated independently to predict the presence of CRC, each producing an independent classification outcome. The confidence levels of these predictions were evaluated.
- Classifier Methods: A meta-classifier was utilized, which took the outputs from the individual modules as inputs to determine the final decision. The fusion process was optimized by tuning the aggregation rules or adjusting the parameters of the meta-classifier
- Performance Testing: The combined system was tested on a diverse dataset that had not been exposed to during the training of the individual modules.
- Fusion Rule Adjustment: The decision-fusion rules were adjusted, if necessary, to improve performance or to handle discrepancies between the predictions of the modules.
- Incorporation of Additional Training Data: In cases of underperformance, the expansion of training datasets was considered, or additional features were included in the models.
- Performance metrics for tuning: Cross-validated accuracy