Durch den Einsatz fortschrittlicher Technologien wie Big Data und Künstlicher Intelligenz (KI) können Gesundheitsdienstleister nicht nur die Diagnosegenauigkeit verbessern, sondern auch einen proaktiven Ansatz ermöglichen, der frühe Interventionen und personalisierte Präventionsmaßnahmen erlaubt und letztlich zu einer besseren Gesundheitsversorgung führt. Die Integration verschiedener medizinischer Datenquellen, wie beispielsweise Patientenfeedback, genetische Daten und elektronische Gesundheitsakten, ermöglicht ein umfassendes Verständnis der einzigartigen Umstände jedes Patienten und kann zudem zur Entdeckung neuer Muster und Korrelationen führen, die die medizinische Forschung und Fortschritte in der Biotechnologie weiter fördern.
Multimodale medizinische Datenfusion
In der multimodalen Technologie kann eine einzelne Bildgebungsmethode die Schwächen einer anderen Methode ergänzen, um den medizinischen Zustand genau zu bewerten und diagnostische Informationen durch die Fusion von Informationen aus mehreren Modalitäten zu erhalten. Multimodale Fusion bezieht sich auf Repräsentationen, und ein Prozess, der sich darauf konzentriert, mithilfe einer Architektur Repräsentationen verschiedener Einzelmodelle zu verschmelzen, wird als Fusion klassifiziert.

HSA-Lösungen für die medizinische Bildgebung
Medizinische Bildgebung ist ein effektives Mittel zur Krankheitsfrüherkennung und zur Lokalisation von Läsionen, aber große Datenmengen bedeuten, dass Fachleute viel Zeit damit verbringen müssen, Analysen durchzuführen und Schlussfolgerungen zu ziehen, die oft subjektiv sind. Der Einsatz von Künstlicher Intelligenz kann nicht nur den Prozess beschleunigen, sondern auch die Wahrscheinlichkeit von Fehldiagnosen verringern. Allerdings gibt es einige Probleme:
- Unterschiedliche Bildqualität: Einige Bilder sind relativ schlecht in der Qualität, mit viel Rauschen, verschwommenen Kanten und einem komplexen und uneinheitlichen Hintergrund, was die Extraktion von Merkmalen und ihre Analyse sehr erschwert.
- Mangelnde Standardisierung: Große Datenmengen erfordern manuelle Annotationen durch Fachleute, den Schutz der Privatsphäre der Patienten, seltene und nicht generalisierte Datenkennzeichnungen.
- Bildungenauigkeit: Medizinische Bilder haben multimodale Bildkanäle mit komplexem Rauschen im Vergleich zur herkömmlichen Segmentierung von natürlichen Bildern. Echtzeit-Leistung und höhere Bildgenauigkeit sind erforderlich.




HS Analysis nutzt Deep-Learning-Algorithmen, die sich besonders gut eignen, die komplexen und heterogenen Datentypen zu verarbeiten, die in der modernen klinischen Praxis gewonnen werden, um die äußerst komplexe Beziehung zwischen Merkmalen und Labels zu erlernen und so Ärzte bei relevanten Analysen und Vorhersagen zu unterstützen.
Digitalisierung von Objektträgern
Das HSA KIT ist extrem vielseitig und lässt sich gut mit einigen der besten automatischen Objektträger-Scanner wie dem Leica GT 450 kombinieren. Es wurde speziell entwickelt, um sich nahtlos mit solchen Geräten zu integrieren und eine schnelle und präzise Objektträger-Scannung zu ermöglichen. Das HSA KIT bietet zudem modernste Funktionen wie automatisierte Objektträger-Handhabung und anpassbare Scan-Parameter, was die Integration in die digitale Pathologie-Infrastruktur weiter verbessert. Benutzer können diese Funktionen nutzen, um ihre Scans zu personalisieren und den Scan-Prozess an ihre Bedürfnisse anzupassen. Fachleute in Disziplinen wie Pathologie und Forschung werden das HSA KIT aufgrund seiner nahtlosen Integration in ihre bestehende Technologie als ausgezeichnete Wahl empfinden.
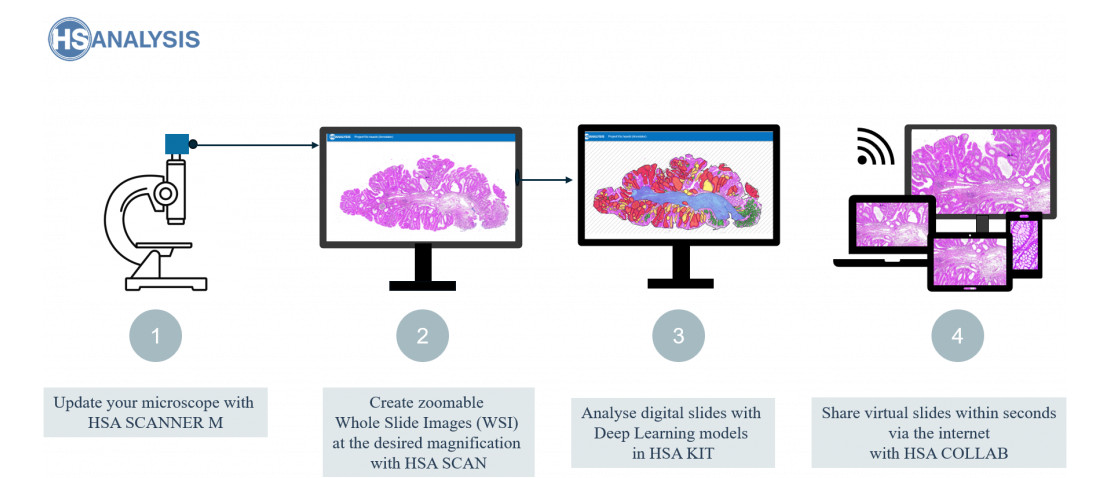
Gleichzeitig bietet HS Analysis eine erschwingliche Alternative zu anderen automatischen Scannern – die HSA Scan M-Software. Die Objektträger können manuell direkt am aktuellen Mikroskop digitalisiert werden, um manuelle WSI (Whole Slide Images) zu erstellen. Dies ermöglicht eine größere Flexibilität und Kontrolle über den Scan-Prozess. Zusätzlich kann das HSA KIT mit einer Vielzahl von Mikroskopmodellen verwendet werden, was es zu einer vielseitigen Option für Labore mit mehreren Mikroskopen macht. Die Software ist benutzerfreundlich und leicht zu navigieren, was ein effizientes Scannen und die Bildverarbeitung ermöglicht.
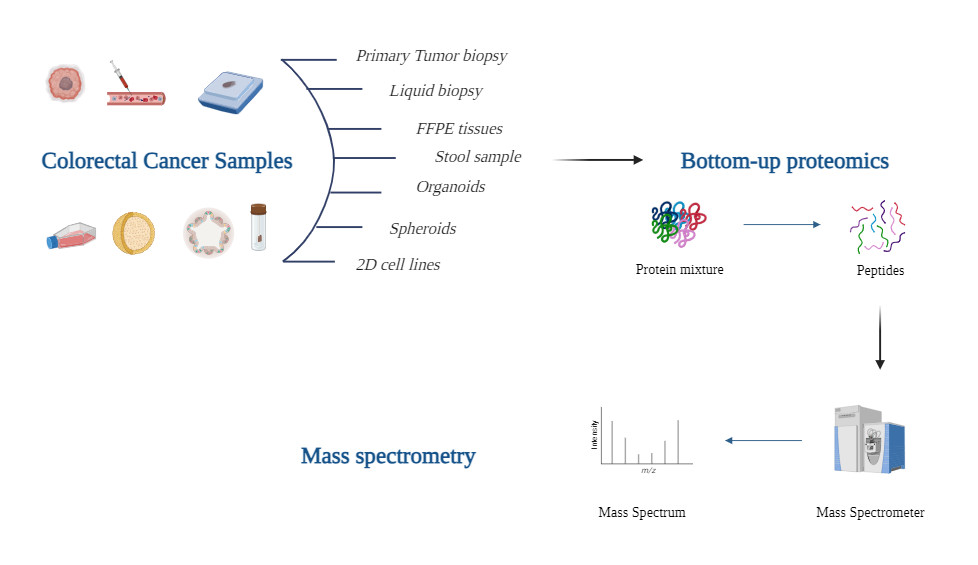
Verwendung von Massenspektrometrie-Daten
Die metabolische Umprogrammierung während der frühesten Stadien des Krebswachstums initiiert eine radikale Veränderung der Zellchemie. Hinweise auf die Onkogenese finden sich in subtilen Veränderungen der Zusammensetzung von Tumoren, wenn sie sich von abnormen Zellen zu aggressiven metastatischen Krebsarten entwickeln. Die Krebsforschung hat zunehmend massenspektrometrie-basierte Profile klinischer Proben (wie Gewebe und Körperflüssigkeiten) übernommen. Eine häufige Strategie besteht darin, Unterschiede im Proteinausdruck zwischen Tumor- und gesundem Gewebe oder zwischen Blut von Kranken und Gesunden zu beschreiben.
MS-Analyse im HSA KIT
Es gibt viele verschiedene Möglichkeiten, MS-Daten im HSA KIT zu visualisieren und zu verstehen. Das Programm kann derzeit .d (Bruker) und .mzML-Dateien nahtlos verarbeiten. Darüber hinaus können die Ergebnisse in verschiedenen Formaten exportiert werden, um sie weiter zu analysieren oder mit anderen Partnern zu teilen. Die Flexibilität des HSA KIT ermöglicht eine nahtlose Integration mit anderen Analysewerkzeugen und Workflows. Neue Dateiformate werden in das Programm aufgenommen, um seine Funktionalität weiter zu verbessern.
Zudem können die integrierten statistischen Analysetools verwendet werden, um Trends und Muster in den Daten zu identifizieren. Die kontinuierlichen Updates und Verbesserungen des HSA KIT stellen sicher, dass die Benutzer Zugriff auf die neuesten Funktionen und Möglichkeiten der MS-Datenanalyse haben. Es ist möglich, Proteomanalysen an Tumorgeweben durchzuführen und die Ergebnisse mit denen von gesundem Gewebe zu vergleichen. Dies kann wertvolle Einblicke in die molekularen Mechanismen der Krebsentstehung und -progression liefern. Darüber hinaus bietet das HSA KIT benutzerfreundliche Schnittstellen und anpassbare Einstellungen, um den individuellen Forschungsanforderungen gerecht zu werden.
MS Viewer: Zum Importieren, Visualisieren und Interpretieren von MS-Daten, bei denen Rohdaten mit FASTA-Dateien kombiniert werden, um detaillierte Informationen über die im Sample identifizierten Proteine zu liefern und Protein-Protein-Interaktionen zu analysieren, um Einblicke in biologische Wege und Netzwerke zu gewinnen.
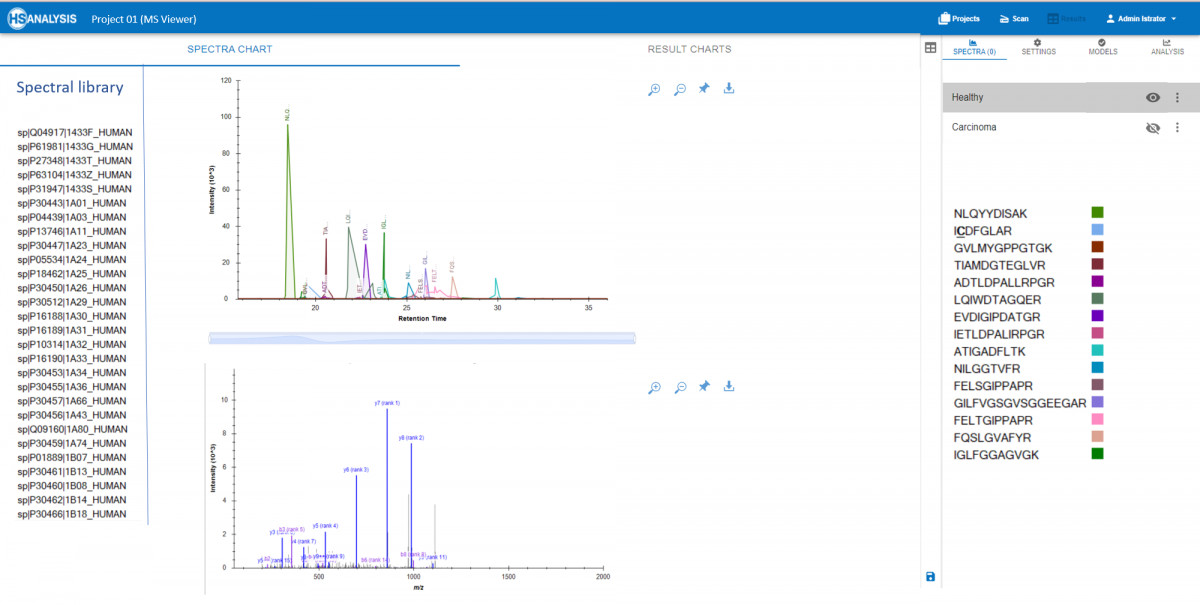
Es besteht die Möglichkeit, Peaks und Spektren von Peptiden in Echtzeit zu betrachten, zusammen mit den entsprechenden Proteinen aus der Spektrenbibliothek. Darüber hinaus ermöglicht das MS-Viewer-Modul den Vergleich mehrerer Proben, um Unterschiede in der Proteinexpression oder -modifikation zu ermitteln. Dieses umfassende Tool ist für Forscher, die wichtige Erkenntnisse aus ihren Massenspektrometrie-Experimenten gewinnen wollen, unerlässlich. Eine Anwendung ist der Vergleich von MS-Peaks in gesundem und krebsartigem Gewebe, wo jedes Fehlverhalten von Peptiden leicht erkannt und weiter analysiert werden kann.
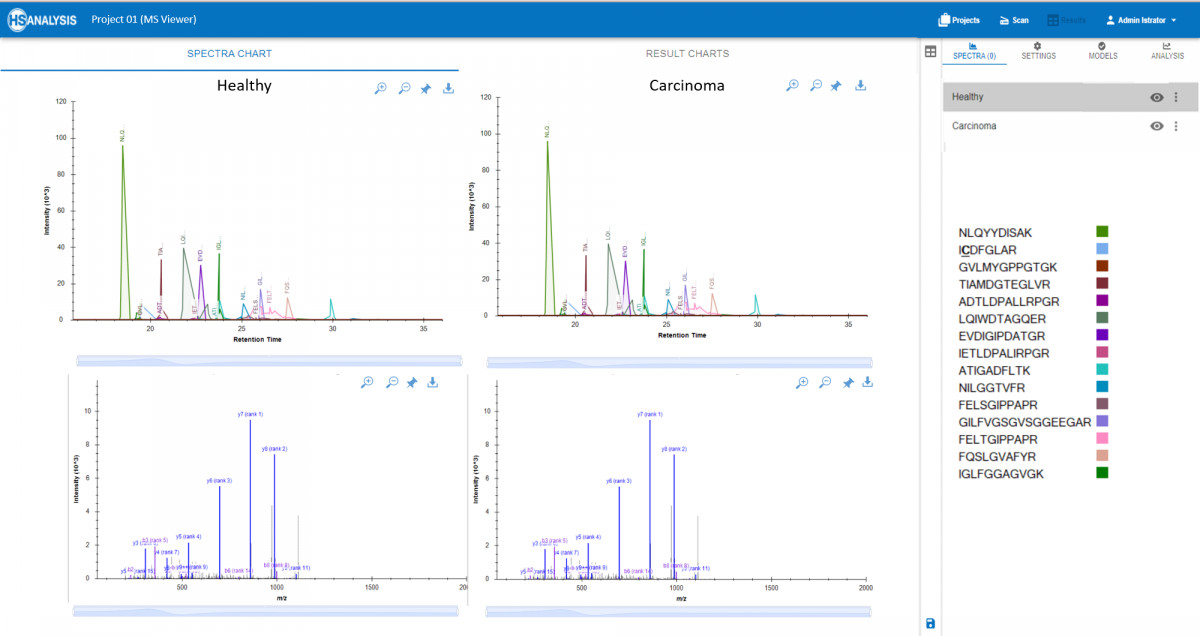
MS Viewer ermöglicht nicht nur die Visualisierung, sondern erstellt am Ende auch einen Bericht, der Informationen über jedes in einem Protein vorhandene Peptid (Vorläufer) mit seiner Häufigkeit und anderen relevanten Faktoren enthält. Zusätzlich besteht die Möglichkeit, eine vergleichende Quantifizierung der Daten zu erhalten, bei der signifikante Unterschiede zwischen zwei Dateien hervorgehoben werden können.
MS Analyser: In Anlehnung an die Ergebnisse von MS Viewer ist dieses Modul ein leistungsstarkes Werkzeug zur Untersuchung von Proteinstrukturen und -funktionen. Der Schwerpunkt liegt auf der Suche nach Peptidpositionen und deren Zuordnung zur gesamten Proteinsequenz, um Proteininteraktionen in Netzwerken besser zu verstehen. Darüber hinaus kann MS Analyser auch zur Bestimmung posttranslationaler Modifikationen von Proteinen verwendet werden, was weitere Einblicke in ihre Funktionen und Regulierungsmechanismen ermöglicht. Durch die Analyse von Peptidpositionen und deren Zuordnung zur Proteinsequenz können Forscher Schlüsselreste aufdecken, die an Proteininteraktionen und Signalwegen beteiligt sind.
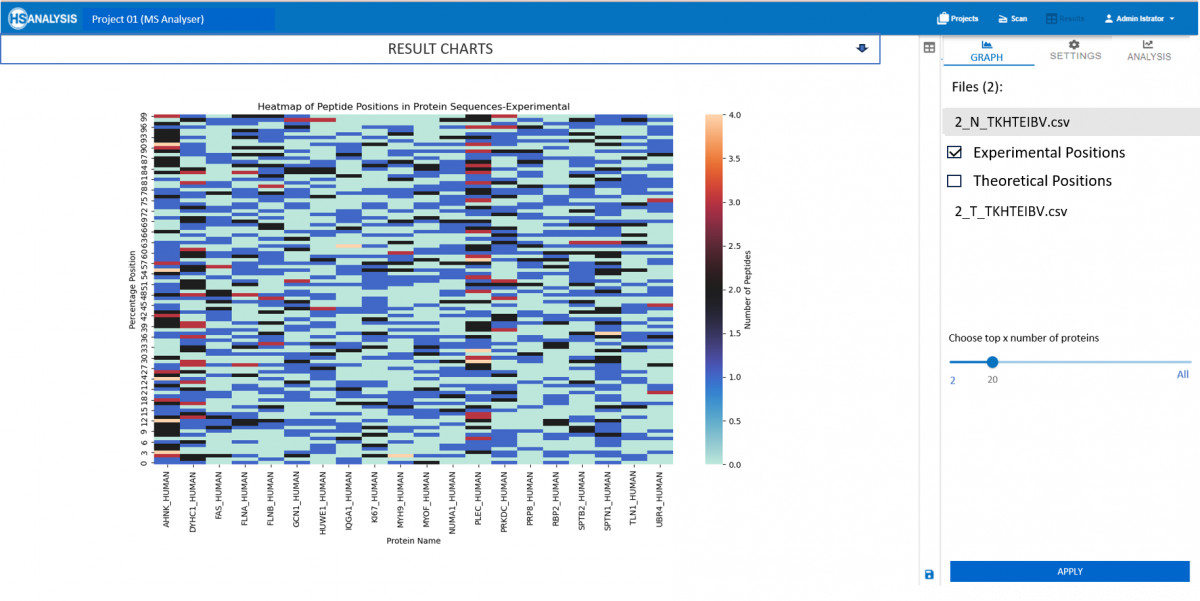
Die Versuchsergebnisse entsprechen der .csv-Eingabedatei, die von unserem MS-Viewer erzeugt wurde. Sie enthält wichtige Informationen über Qualität und Quantität der Peptide. Diese Daten können dann mit Hilfe von Bioinformatik-Tools weiter analysiert werden, um Muster und Beziehungen zwischen Proteinen zu erkennen. Durch die Integration mehrerer Datenquellen lässt sich ein umfassendes Verständnis der Proteinfunktion und -regulierung in biologischen Systemen gewinnen.
Bei den theoretischen Ergebnissen handelt es sich um das erwartete Verhalten von Peptiden, wenn ihr jeweiliges Protein mit Trypsin verdaut wird. Dies gibt wertvolle Einblicke in die erwartete Proteinstruktur und ihr Verhalten. Darüber hinaus können diese Daten zur Validierung von Versuchsergebnissen und zur Festlegung künftiger Forschungsrichtungen im Bereich der Proteomik verwendet werden.
Der praktikabelste Anwendungsfall dieses Moduls ist die Überprüfung, welche Peptide fehlen oder in höherer Konzentration vorliegen als in den experimentellen Ergebnissen erwartet.
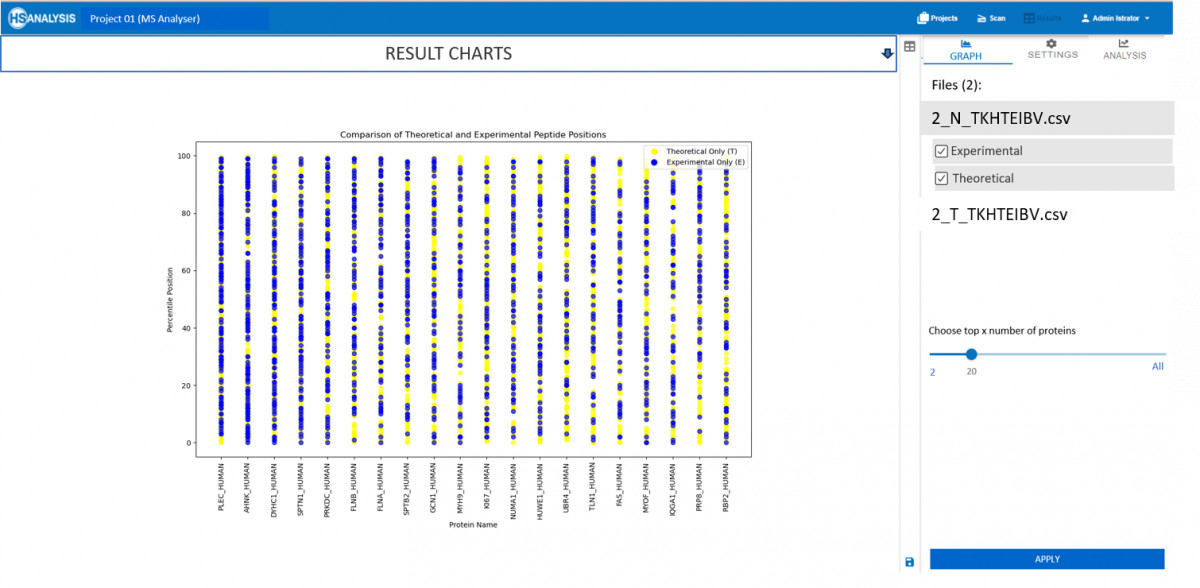
Nachdem nun die tatsächlichen und die erwarteten Peptidpositionen ermittelt wurden, kann auf der Registerkarte „Analyse“ automatisch eine detaillierte Übersicht angezeigt werden. Besonders wichtig ist, dass der Vergleich dieser Art von Ergebnissen auch für zwei verschiedene Dateien möglich ist, z. B. für gesundes und krebsartiges Gewebe.
Zusammenfassend lässt sich sagen, dass MS Viewer in erster Linie ein Werkzeug zur Untersuchung der Proteinexpression ist, während MS Analyser sich mit der Position der Peptide befasst. Eine Kombination dieser beiden Module ergibt eine End-to-End-Lösung für die Proteomik, insbesondere für Krebsstudien, bei denen große Datenmengen in einfachster Form interpretiert werden müssen.
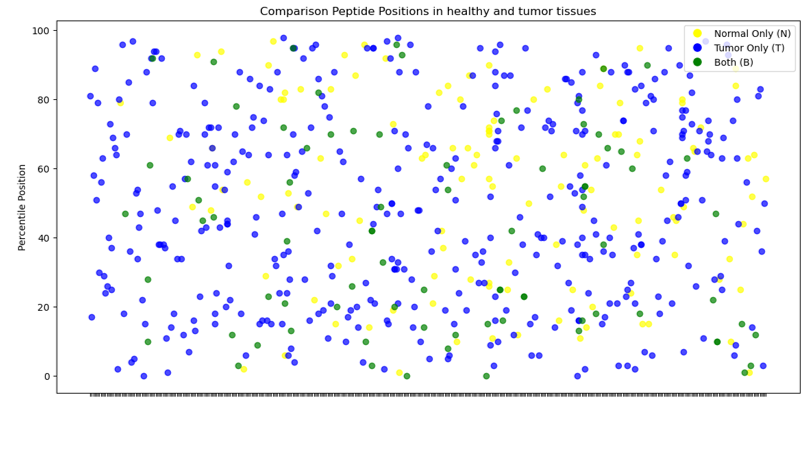
Zusammenfassend lässt sich sagen, dass das MS-Analysegerät in der Lage ist, zwischen normalem und Tumorgewebe auf molekularer Ebene zu unterscheiden, wobei Unterschiede in der Protein- und Peptidzahl festgestellt wurden. Einige Proteine weisen eine höhere Peptidzahl auf, auch wenn dies nicht in allen Fällen von CRC der Fall ist. Schließlich deuten grafische Darstellungen darauf hin, dass bestimmte Peptide in Tumorgeweben im Vergleich zu normalem Gewebe fehlen, was auf spezifische molekulare Veränderungen im Zusammenhang mit Krebsumwandlungen hindeutet.
Die Häufigkeit der Peptide allein kann nicht viel über ihr Verhalten aussagen, es sei denn, sie wird zusammen mit ihrer Intensität untersucht. Theoretisch müssen die Peptide, die zu demselben Protein gehören, die gleiche Stöchiometrie aufweisen, d. h. sie müssen in gleichen molaren Mengen vorhanden sein. Ihre Erkennung in Massenspektrometern wird jedoch durch das Vorhandensein anderer Vorläufer-Ionen beeinflusst.

Forschung ist ein zeitaufwändiger Prozess, der langfristige Antworten erfordert. Die Verwendung solcher Bibliotheken schränkt die Fähigkeit zur Integration in künftige Bestrebungen ein. Darüber hinaus kann der ausschließliche Rückgriff auf bereits bestehende Bibliotheken die Fähigkeit einschränken, sich an neue Technologien oder Ansätze, die in der Zukunft auftauchen, anzupassen und diese zu integrieren. Für Forscher und Entwickler ist es von entscheidender Bedeutung, dass sie mit neuen Methoden experimentieren und ihre Lösungen an die sich ändernden Forschungsanforderungen anpassen können.
HSA KIT bietet eine nahtlose Integration in bestehende Laborgeräte und Softwaresysteme, wodurch der Arbeitsablauf rationalisiert und die Effizienz maximiert wird. Dies ermöglicht es Forschern und Entwicklern, sich mehr auf ihre wissenschaftlichen Ziele als auf technische Herausforderungen zu konzentrieren.
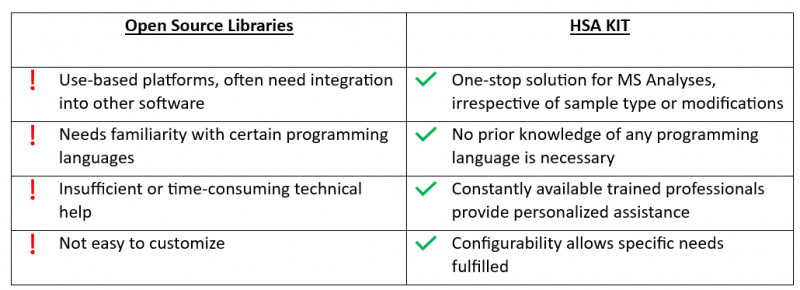
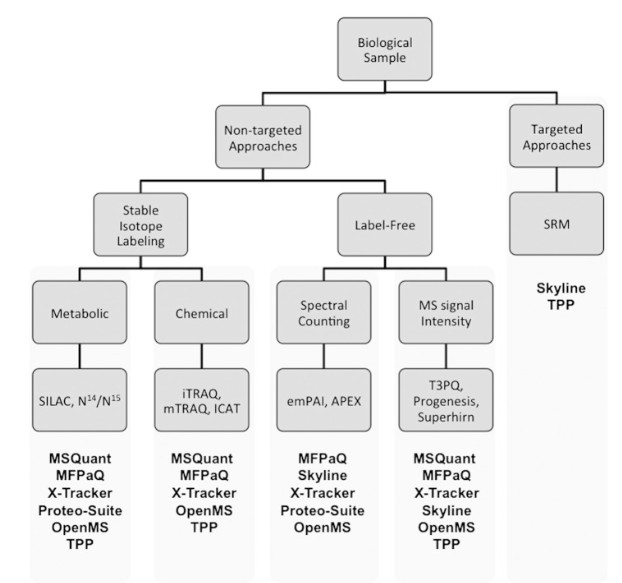
Eine Gemeinschaft von Bioinformatikern und Softwareentwicklern entwickelt und pflegt eine breite Palette von Softwarelösungen, die die meisten Elemente der MS-Datenanalyse abdecken.
Zu den üblichen Aufgaben der MS-Datenverarbeitung gehören die theoretische Proteomanalyse, die Verarbeitung von Rohspektren, die Konvertierung von Dateiformaten, die Erstellung von Identifikationsstatistiken und die Speicherung/Visualisierung von Rohdaten, Identifikations- und Quantifizierungsergebnissen.

Open-Source-Software hat gewisse Nachteile, wie z. B. eine begrenzte Kompatibilität mit bestimmten Datenformaten, eine schwierige Benutzeroberfläche und einen Mangel an gründlicher technischer Unterstützung.
HSA KIT vereinfacht nicht nur den Analyseprozess für die Massenspektrometrie, sondern bietet auch eine anpassungsfähige Plattform für die Einbeziehung von Deep-Learning-Techniken. Dies erweitert sein Potenzial über konventionelle Massenspektrometrieanwendungen hinaus, indem es die Modifizierung von Modulen für eine Vielzahl von Anwendungen wie Krankheitserkennung, Zertifizierung von Lebensmitteltoxizität oder Hauptkomponentenanalyse ermöglicht. Ganz zu schweigen von der Fähigkeit, mit verschiedenen Datentypen zu arbeiten und bei Bedarf automatische Dateikonvertierungen durchzuführen.
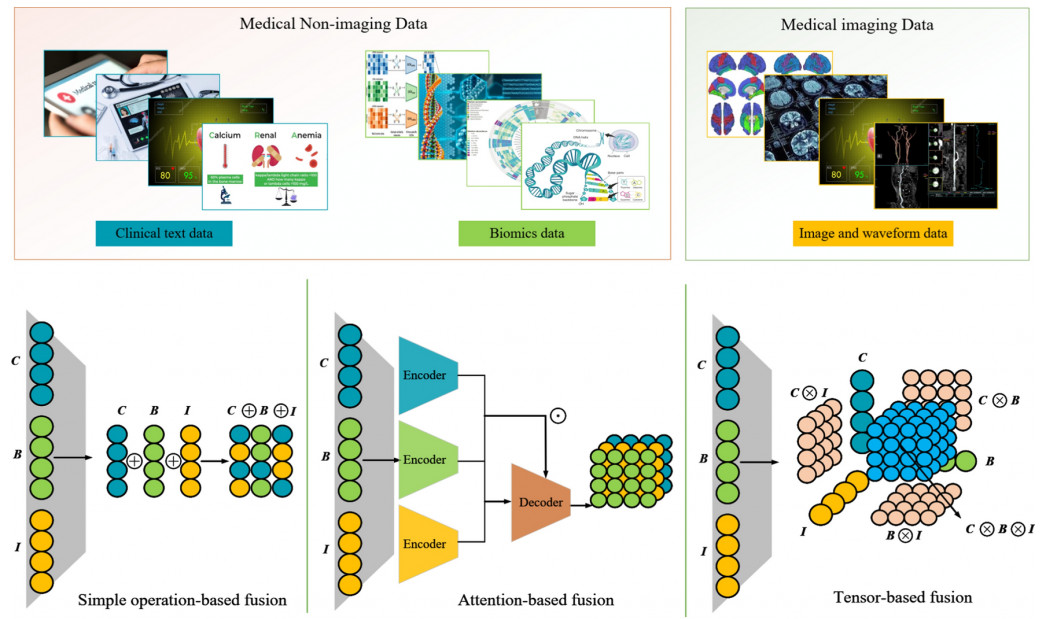
Medizinische Bildfusion
Multimodale medizinische Bildfusion, die auf Deep Learning basiert, kann verwendet werden, um die Merkmalsinformationen verschiedener Modi effektiv zu extrahieren und zu integrieren, die klinische Anwendbarkeit von medizinischen Bildern bei der Diagnose und Bewertung von medizinischen Problemen zu verbessern sowie eine quantitative Analyse, Echtzeitüberwachung und Behandlungsplanung für Ärzte und Forscher zu ermöglichen.
Derzeit gibt es zwei Hauptansätze für multimodale Aufgaben: leichte Fusion und schwere Fusion.
- Leichte Fusion: normalerweise mühelos, wie das Vektor-Innenprodukt, dargestellt durch CLIP und ALIGN, die eine Zwei-Turm-Struktur verwenden, die sich auf die multimodale Ausrichtung konzentriert, um Aufgaben wie Textabgleich, -abruf und andere nachgelagerte Aufgaben zu erleichtern
- Schwere Fusion: basiert auf vortrainierten Transformern, wie sie durch OSCAR, UNITER, VINVL usw. repräsentiert werden. Schwere Fusion kann VQA, Bildunterschriften und andere nachgelagerte Aufgaben interpretieren, die eine Informationsfusion und -verständnis erfordern, was der ALIGN-Algorithmus nicht leisten kann
Fusionsmethoden können je nach ihrem Einsatzort in späte und frühe Fusion unterteilt werden.
- Frühe Fusion: Ihr Vorteil liegt darin, früh in der Analyse Korrelationen zwischen verschiedenen Merkmalen aus mehreren Modalitäten zu nutzen. Wenn eine starke Korrelation zwischen den Modalitäten besteht, wie beim Zusammenführen von Audio- und visuellen Eingaben für die Spracherkennung, wird diese Methode empfohlen
- Späte Fusion: Jede Modalität muss ihre eigenen Modelle trainieren, und die Ergebnisse müssen dann kombiniert werden. Nach dem Kombinieren von Merkmalsvektoren aus verschiedenen unabhängigen Quellen trifft die späte Fusion Entscheidungen auf Basis der gemeinsamen Merkmale, die daraus entstehen
- Hybride Fusion: optimiert die Vorteile der frühen und späten Fusion. Sie erhöht jedoch auch die Komplexität des Modells und erschwert das Training. In Situationen, in denen Flexibilität erforderlich ist, könnten hybride Fusionsansätze aufgrund der Anpassungsfähigkeit von Deep-Learning-Modellstrukturen die bessere Wahl sein
Es gibt drei Methoden zur Fusion von Text und Bild: einfache operationbasierte, auf Aufmerksamkeit basierende und tensorbasierte Ansätze.
Hybride Architektur von CNN/RNN
CNNs sind eine Familie von diskriminativen Modellen, die in den meisten Anwendungen der Computer Vision, einschließlich Objekterkennung, semantischer Segmentierung, Bildklassifizierung und so weiter, ihre Wirksamkeit gezeigt haben. Sie nutzen die räumlichen Merkmale visueller Eingabeströme. RNN-Netzwerke sind besser geeignet, die zeitlichen Zusammenhänge komplexer Aufgaben wie Spracherkennung und maschinelle Übersetzung zu reflektieren, da sie sequentielle Daten und Zeitreihen besser modellieren können als CNN. Anders ausgedrückt: RNN ist besser darin, Korrelationen in raumzeitlichen Informationen zu berücksichtigen, während CNN effektiver visuelle Merkmale aus statischen Daten wie Bildern, Tiefenkarten usw. extrahiert. Für eine Vielzahl von Anwendungen wurden Versuche unternommen, CNN- und RNN-Architekturen zu kombinieren. Noch spezifischer: RNN sagt den nächsten Zustand in der beobachteten Ereignissequenz basierend auf zuvor verborgenen Zuständen voraus, während CNN Faltungskerne verwendet, um räumliche Daten zu transformieren.
Multi-Modal Analyser (MMA) im HSA KIT
Die Fusion von zwei Modalitäten in der CRC-Studie, Bildanalyse und Proteomsequenzanalyse, wurde unter Verwendung einer Fusionsmethode auf Entscheidungsebene durchgeführt. Zwei separate Module wurden unabhängig voneinander trainiert und validiert, um das Vorhandensein von CRC vorherzusagen, wobei jedes Modul ein unabhängiges Klassifizierungsergebnis lieferte. Die Vertrauensniveaus dieser Vorhersagen wurden bewertet.
- Klassifiziermethoden: Ein Meta-Klassifizierer wurde verwendet, der die Ergebnisse der einzelnen Module als Eingaben nutzte, um die endgültige Entscheidung zu treffen. Der Fusionsprozess wurde durch Feinabstimmung der Aggregationsregeln oder durch Anpassung der Parameter des Meta-Klassifizierers optimiert.
- Leistungstests: Das kombinierte System wurde auf einem vielfältigen Datensatz getestet, der während des Trainings der einzelnen Module nicht verwendet wurde.
- Anpassung der Fusionsregeln: Die Entscheidungsfusionsregeln wurden, falls erforderlich, angepasst, um die Leistung zu verbessern oder um Unstimmigkeiten zwischen den Vorhersagen der Module zu beheben.
- Einbeziehung zusätzlicher Trainingsdaten: Bei unzureichender Leistung wurde die Erweiterung der Trainingsdatensätze in Betracht gezogen oder zusätzliche Merkmale in die Modelle einbezogen.
- Leistungskennzahlen zur Feinabstimmung: Kreuzvalidierte Genauigkeit