Bei HS Analysis spezialisieren wir uns auf die Zukunft der Krebsforschung mit Deep Learning. Wir stehen an der Spitze der Integration von Spitzentechnologie und Lebenswissenschaften. Unsere neueste Innovation schlägt eine Brücke zwischen Proteomik und Onkologie und bietet bahnbrechende Werkzeuge für die wissenschaftliche Gemeinschaft.
Die Krebsforschung nutzt zunehmend massenspektrometrie-basierte Profile klinischer Proben (wie Gewebe und Bioflüssigkeiten). Eine häufige Strategie ist es, Unterschiede im Proteinausdruck zwischen Tumor- und gesundem Gewebe oder zwischen Blut von Kranken und Gesunden zu beschreiben.
HSA KIT: Das Toolkit, das die MS-Datenanalyse revolutioniert
Unser Flaggschiffprodukt, HSA KIT, bietet vielseitige Lösungen zur Visualisierung und zum Verständnis von Massenspektrometrie (MS)-Daten. Kompatibel mit .d (Bruker)- und .mzML-Dateien vereinfacht HSA KIT den Umgang mit Daten und sorgt für eine nahtlose Integration mit anderen analytischen Tools und Workflows. Regelmäßige Updates erweitern die Funktionalität und stellen sicher, dass die Benutzer von den neuesten Funktionen und Möglichkeiten der MS-Datenanalyse profitieren.
Spec2PepNet (MS Viewer): Entschlüsselung von Massenspektrometriedaten
Die Reise beginnt mit unserem fortschrittlichen Deep Learning-Modul, Spec2PepNet. Dieses leistungsstarke Tool wurde entwickelt, um Rohdaten der Massenspektrometrie zu analysieren und sie mit FASTA-Dateien zu kombinieren, um detaillierte Informationen über die in Ihren Proben identifizierten Proteine zu liefern. Zu den wichtigsten Funktionen gehören:
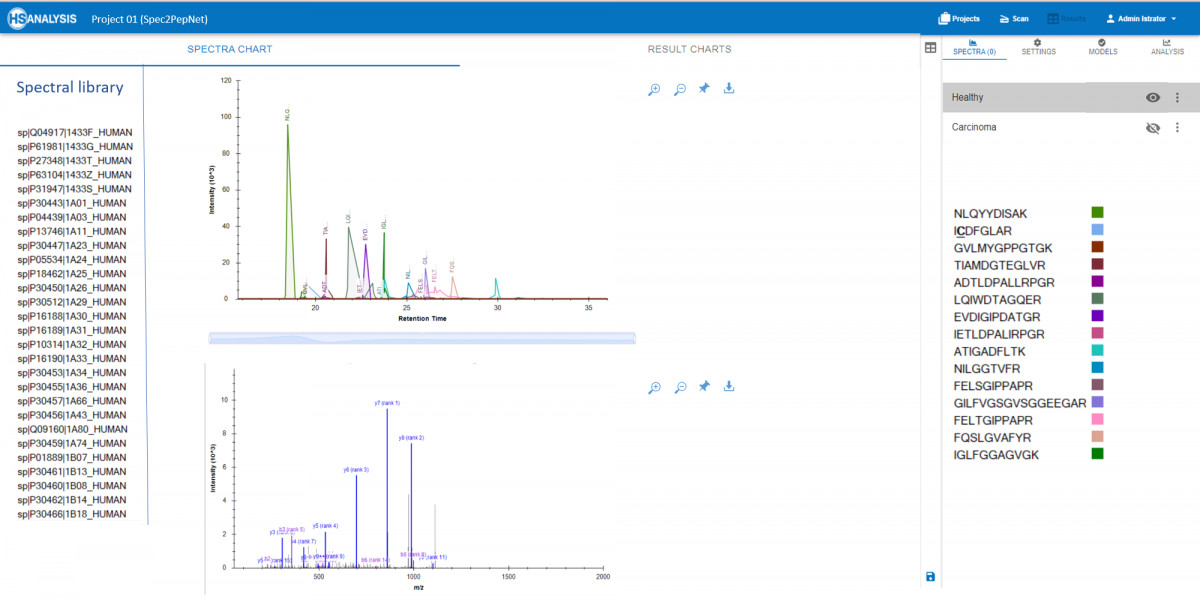
- Echtzeitvisualisierung: Zeigen Sie Peptide und Spektren von Proteinen aus der Spektralbibliothek an.
- Analyse von Protein-Interaktionen: Erhalten Sie Einblicke in biologische Signalwege und Netzwerke durch detaillierte Studien zu Protein-Protein-Interaktionen.
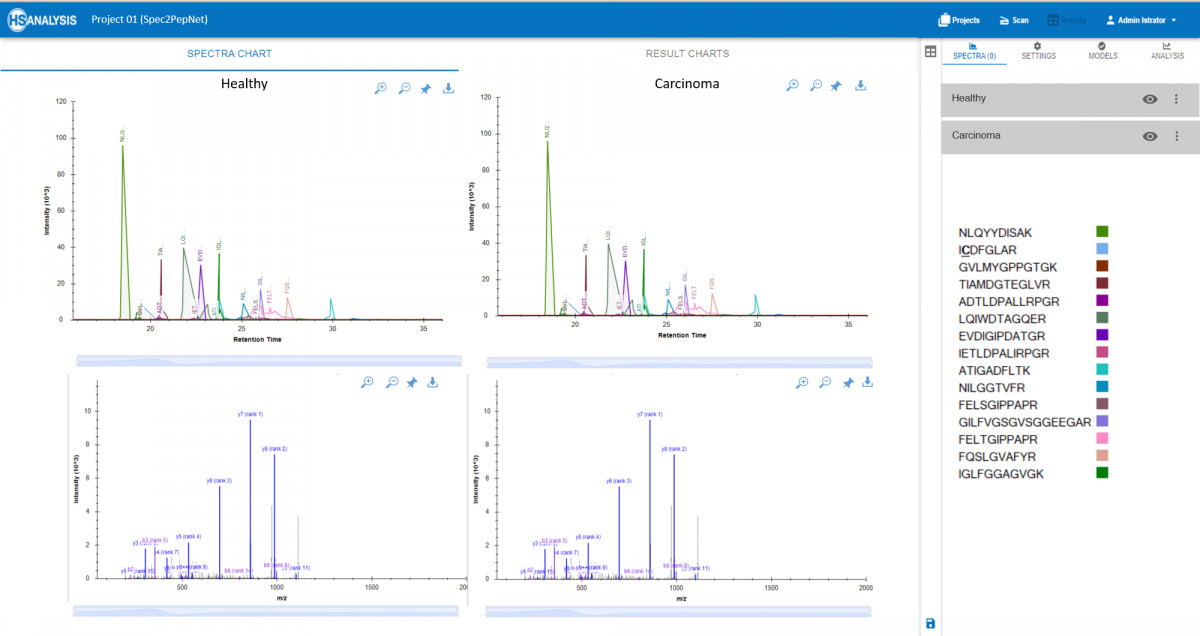
- Vergleich von Proben: Vergleichen Sie mehrere Proben, um Unterschiede in den Proteinausdrucksniveaus oder Modifikationen zu identifizieren, die wesentliche Erkenntnisse in der Krebsforschung liefern.
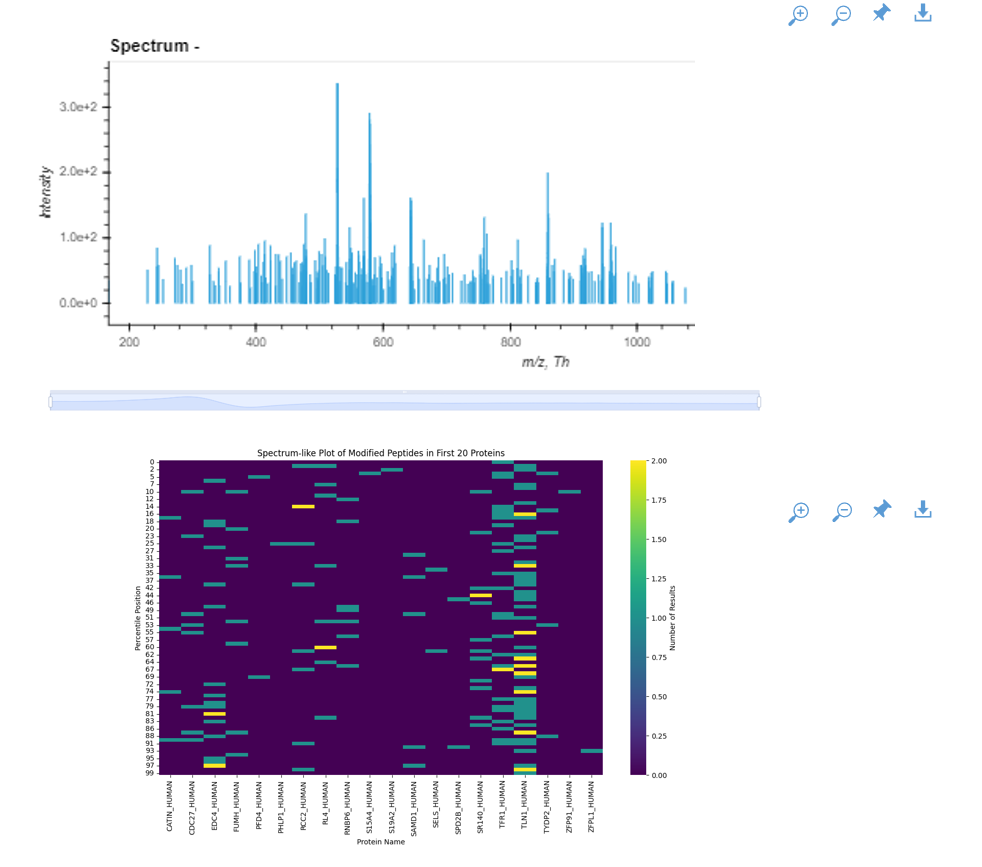
Spec2PepNet erstellt außerdem detaillierte Berichte, die Informationen über die Menge jedes Peptids und andere Faktoren enthalten. Quantitative Vergleiche heben signifikante Unterschiede zwischen Proben hervor, wie z. B. gesundes vs. krebsartiges Gewebe.
MS Analyser: Tiefe Einblicke aus vorhersagbaren Daten
Nach der detaillierten Analyse durch Spec2PepNet übernimmt das Modul MS Analyser das Kommando. Es konzentriert sich auf die Untersuchung von Proteinstrukturen und -funktionen und ermöglicht eine umfassende Untersuchung von Peptidpositionen, indem sie auf ganze Proteinsequenzen abgebildet werden. Zu den wichtigsten Fähigkeiten gehören:
- Peptidkartierung: Vergleichen Sie experimentelle Ergebnisse mit theoretischen Erwartungen, um Erkenntnisse zu bestätigen und künftige Forschungen zu leiten.
- Posttranslationale Modifikationen: Bestimmen Sie diese Modifikationen, um tiefere Einblicke in Proteinfunktionen und regulatorische Mechanismen zu erhalten.
- Netzwerkanalyse: Verstehen Sie Proteininteraktionen in Netzwerken und Signalwegen.
Durch die Analyse der entscheidenden Daten aus der von Spec2PepNet erstellten .csv-Datei entdeckt der MS Analyser Muster und Beziehungen zwischen Proteinen und liefert einen umfassenden Überblick über die Proteinfunktion und -regulation innerhalb biologischer Systeme.
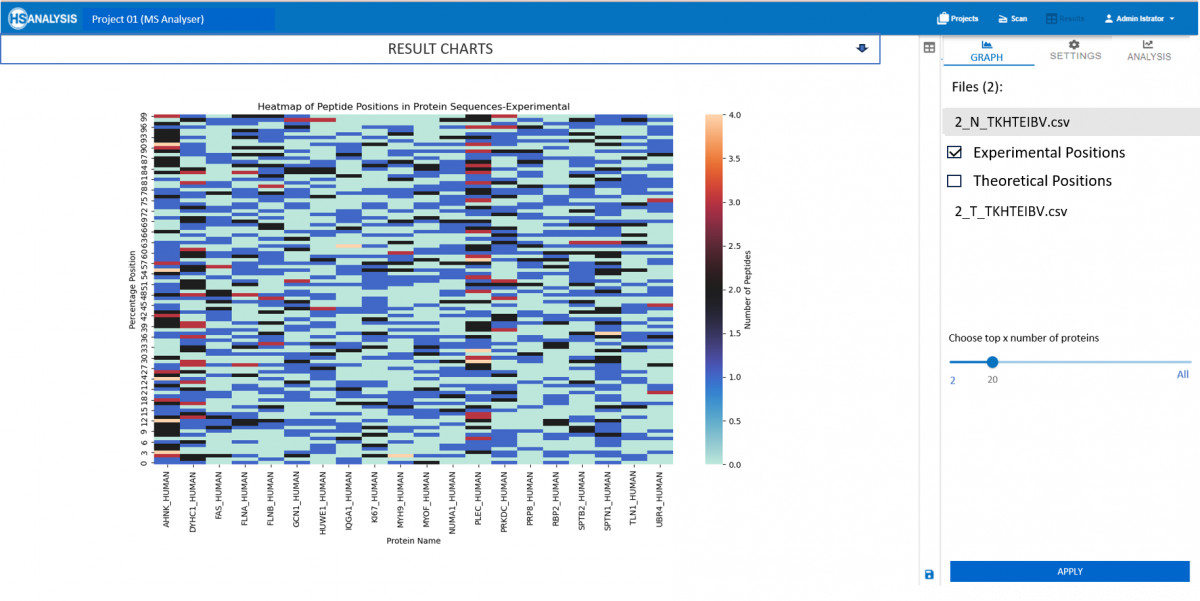
Experimentelle Ergebnisse entsprechen der .csv-Datei, die von unserem MS Viewer erstellt wurde. Sie enthält wichtige Informationen über die Qualität und Quantität der Peptide. Diese Daten können dann mit bioinformatischen Werkzeugen weiter analysiert werden, um Muster und Beziehungen zwischen Proteinen zu identifizieren. Durch die Integration mehrerer Datenquellen kann ein umfassendes Verständnis der Proteinfunktion und -regulation in biologischen Systemen gewonnen werden.
Die theoretischen Ergebnisse stellen das erwartete Verhalten von Peptiden dar, sobald das jeweilige Protein mit Trypsin verdaut wird. Dies liefert wertvolle Einblicke in die erwartete Proteinstruktur und ihr Verhalten. Darüber hinaus können diese Daten verwendet werden, um experimentelle Ergebnisse zu validieren und zukünftige Forschungsrichtungen im Bereich der Proteomik zu leiten.
Der wichtigste Anwendungsfall dieses Moduls besteht darin, zu überprüfen, welche Peptide in experimentellen Ergebnissen fehlen oder in höheren Konzentrationen als erwartet vorhanden sind.
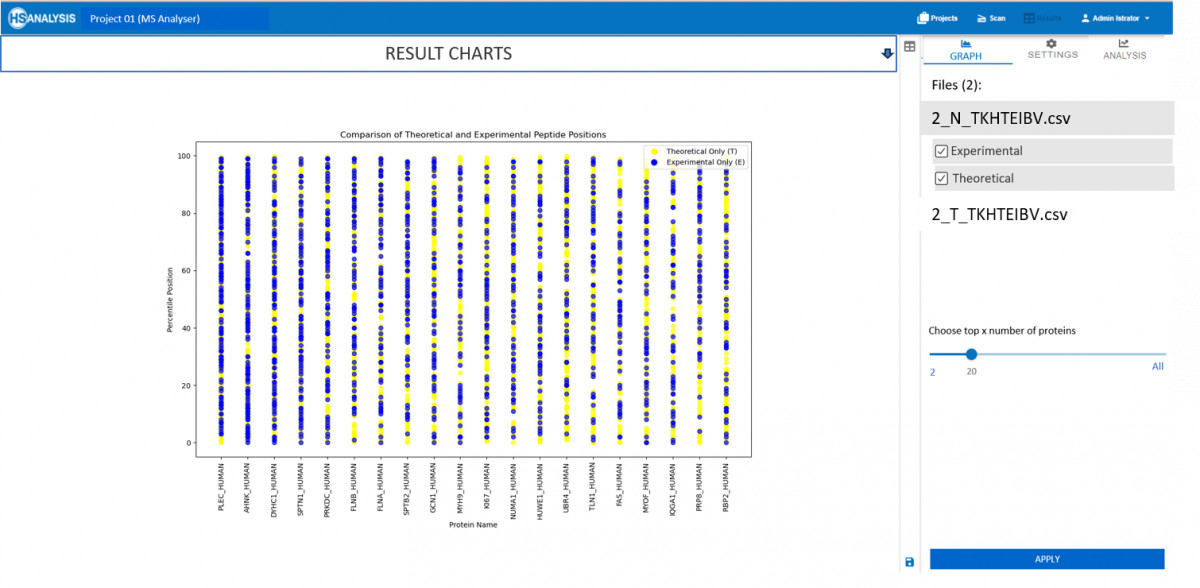
Nun, da die tatsächlichen und erwarteten Peptidpositionen erhalten wurden, kann eine detaillierte Übersicht automatisch im Analyse-Tab gefunden werden. Am wichtigsten ist, dass dieser Vergleichstyp für zwei verschiedene Dateien weiter verglichen werden kann, z.B. gesundes vs. krebsartiges Gewebe.
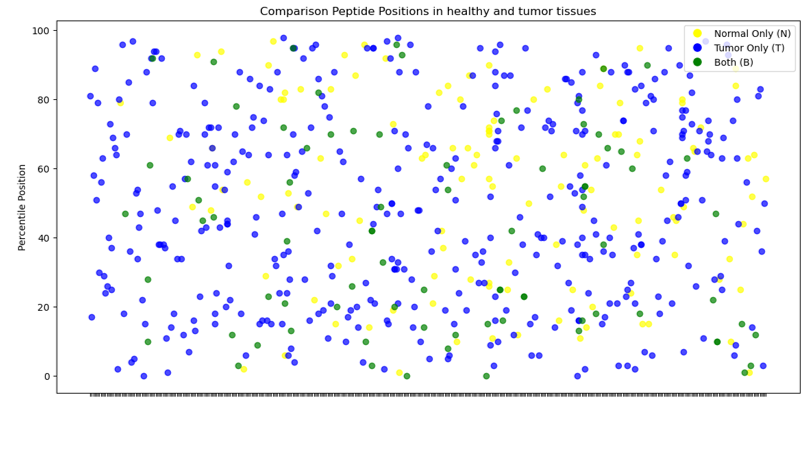
Zusammenfassend lässt sich sagen, dass das MS-Analysegerät in der Lage ist, zwischen normalem und Tumorgewebe auf molekularer Ebene zu unterscheiden, wobei Unterschiede in der Protein- und Peptidzahl festgestellt wurden. Einige Proteine weisen eine höhere Peptidzahl auf, auch wenn dies nicht in allen Fällen von CRC der Fall ist. Schließlich deuten grafische Darstellungen darauf hin, dass bestimmte Peptide in Tumorgeweben im Vergleich zu normalem Gewebe fehlen, was auf spezifische molekulare Veränderungen im Zusammenhang mit Krebsumwandlungen hindeutet.
Die Häufigkeit der Peptide allein kann nicht viel über ihr Verhalten aussagen, es sei denn, sie wird zusammen mit ihrer Intensität untersucht. Theoretisch müssen die Peptide, die zu demselben Protein gehören, die gleiche Stöchiometrie aufweisen, d. h. sie müssen in gleichen molaren Mengen vorhanden sein. Ihre Erkennung in Massenspektrometern wird jedoch durch das Vorhandensein anderer Vorläufer-Ionen beeinflusst.
Führend in der onkologischen Proteomik
Unser Projekt ist ein Beweis für die Relevanz der Proteomik in der Krebsforschung. Durch die Nutzung dieser fortschrittlichen Deep Learning-Modelle treibt HS Analysis eine neue Ära voran, in der die Vorhersage und Interpretation proteomischer Daten zu umsetzbaren Erkenntnissen in der Onkologie führen kann. Wir sind überzeugt, dass dieses Projekt nicht nur die wichtige Rolle der Proteomik unterstreicht, sondern auch Fortschritte fördert, die uns effektiven Krebsbehandlungen näher bringen.
Begleiten Sie uns auf dieser innovativen Reise, um die Krebsforschung durch Proteomik und Deep Learning zu revolutionieren. Entdecken Sie, wie HS Analysis Fortschritte in Richtung einer Zukunft macht, in der Präzisionsmedizin und personalisierte Behandlungen in greifbare Nähe rücken.
Erfahren Sie mehr über unsere Vision und unsere wegweisenden Arbeiten auf HS Analysis
Forschung ist ein zeitaufwändiger Prozess, der langfristige Antworten erfordert. Die Nutzung solcher Bibliotheken schränkt die Fähigkeit ein, zukünftige Ambitionen zu integrieren. Darüber hinaus kann sich das ausschließliche Verlassen auf vorhandene Bibliotheken als Hindernis erweisen, um sich an neue Technologien oder Ansätze anzupassen, die in der Zukunft aufkommen könnten. Es ist für Forscher und Entwickler von entscheidender Bedeutung, neue Methoden ausprobieren und ihre Lösungen an die sich ändernden Forschungsbedürfnisse anpassen zu können.
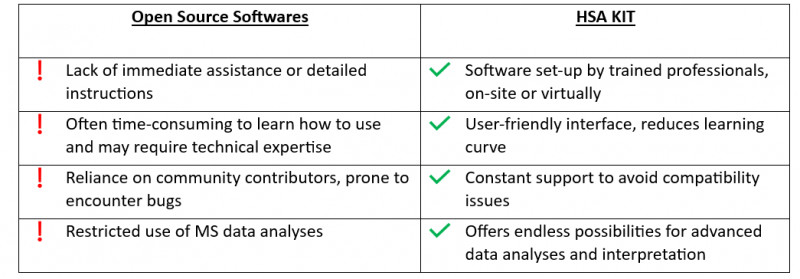
Open-Source-Software hat gewisse Nachteile, wie z.B. eine begrenzte Kompatibilität mit bestimmten Datenformaten, eine schwierige Benutzeroberfläche und mangelnde umfassende technische Unterstützung.
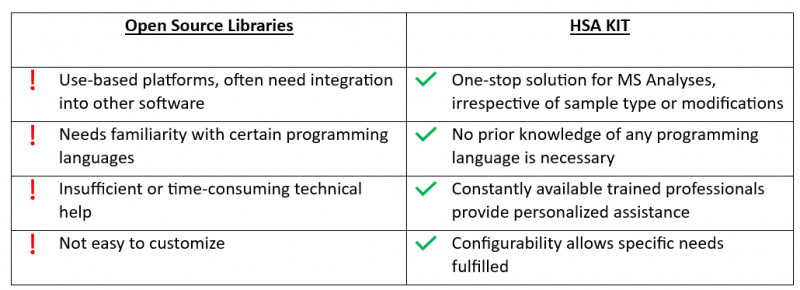
HSA KIT bietet eine nahtlose Integration mit bestehenden Laborausrüstungen und Softwaresystemen, optimiert den Workflow und maximiert die Effizienz. Dies ermöglicht es Forschern und Entwicklern, sich mehr auf ihre wissenschaftlichen Ziele zu konzentrieren, anstatt auf technische Herausforderungen.