At HS Analysis, we specialise in unveiling the future of cancer research with Deep Learning. We are at the forefront of integrating cutting-edge technology with life sciences. Our latest innovation bridges the realm of proteomics and oncology, providing groundbreaking tools for the scientific community.
Proteomics in Cancer research increasingly relies on mass spectrometry-based profiling of clinical specimens. This technique helps identify differences in protein expression levels, offering insights into disease mechanisms and potential therapeutic targets.
HSA KIT: The Toolkit Revolutionizing MS Data Analysis
Our flagship product, HSA KIT, offers versatile solutions for visualizing and understanding Mass Spectrometry (MS) data. Compatible with .d (Bruker) and .mzML files, HSA KIT simplifies data handling and ensures seamless integration with other analytical tools and workflows. Regular updates enhance its functionality, ensuring users benefit from the latest features and capabilities in MS data analysis.
- Unmatched Precision: Analyze complex biological samples with unparalleled accuracy.
- Comprehensive Profiling: Capture and quantify thousands of peptides and proteins in a single run.
- Accelerate Discoveries: Facilitate rapid hypothesis testing and validation in cancer studies.
With regular updates, HSA KIT ensures that users benefit from the latest advancements in MS data analysis, allowing them to stay ahead in their research endeavors.
Key Features:
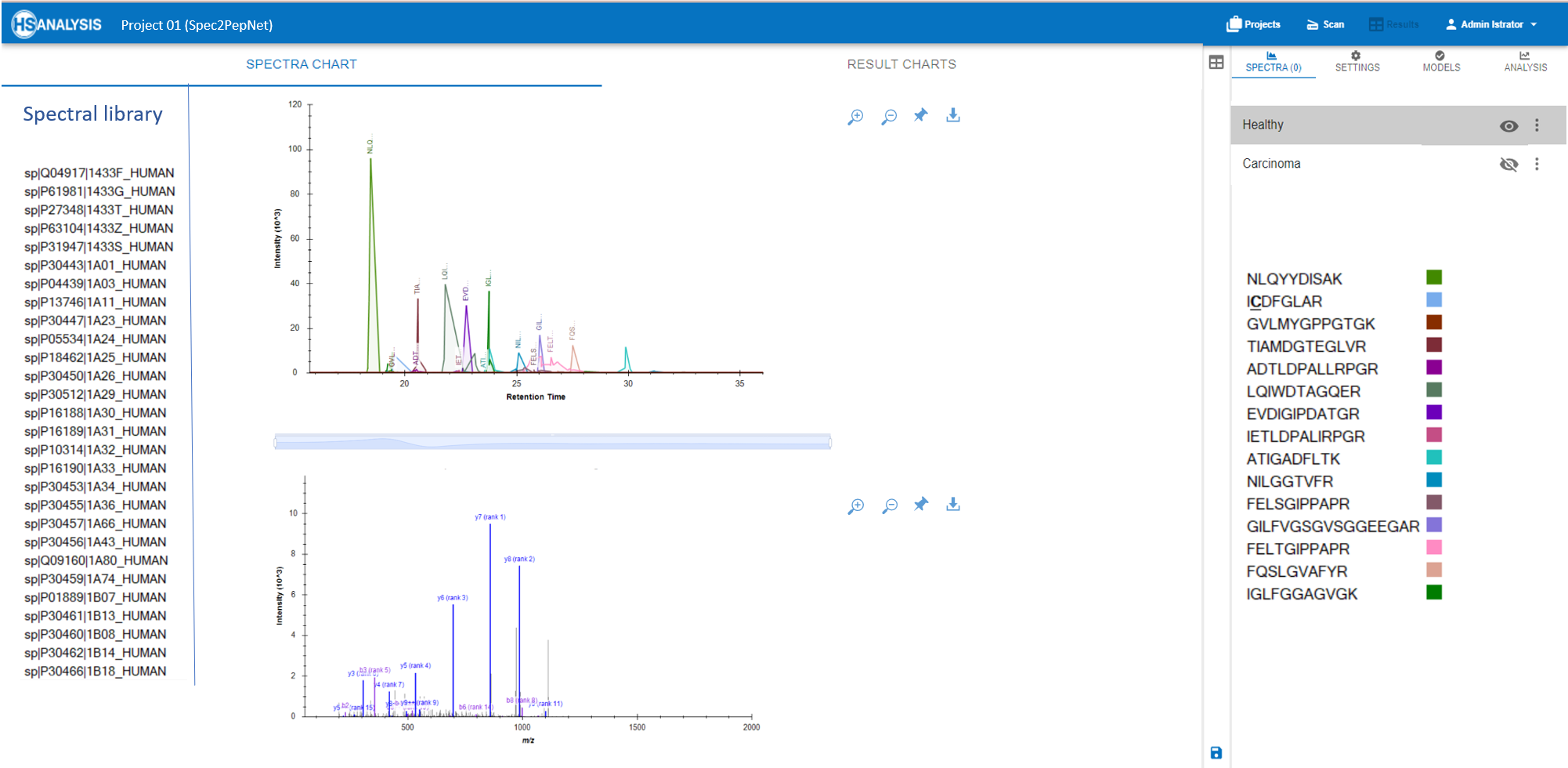
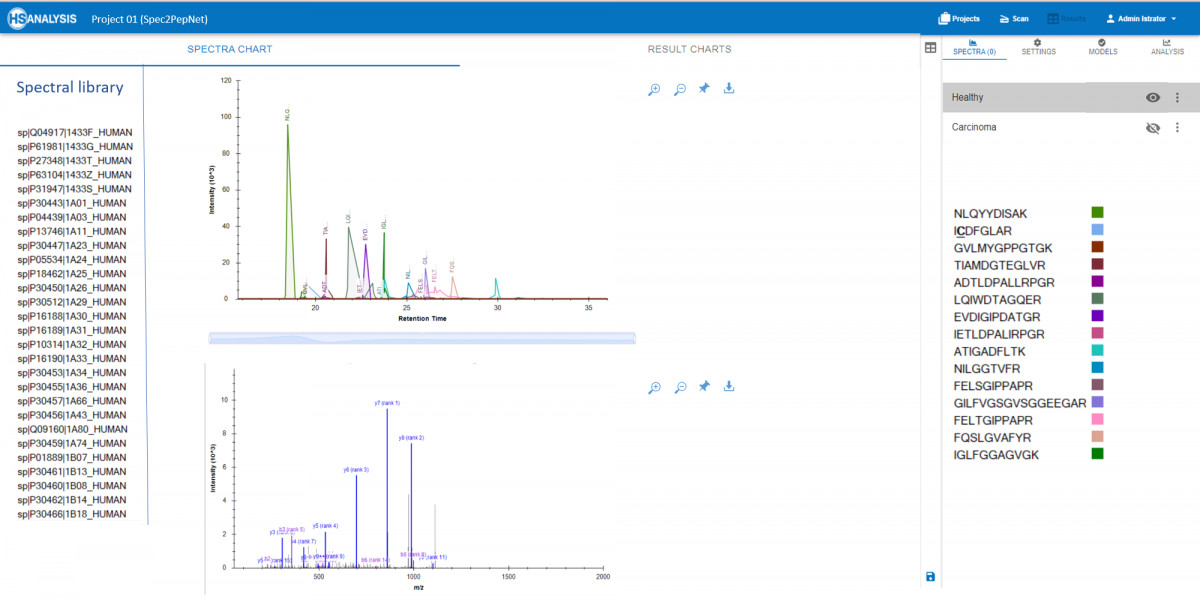
- Real-time Visualization: Interactive visualization tools enable researchers to explore peaks and spectra of peptides alongside corresponding proteins from spectral libraries. This capability aids in quickly identifying patterns and anomalies within datasets.
- Protein Interaction Analysis: Understand intricate biological pathways by mapping protein interactions, helping to elucidate molecular mechanisms underlying oncogenesis and other diseases.
- Sample Comparison: Effortlessly compare multiple samples to detect variations in protein expression levels or post-translational modifications, which are critical for discovering novel insights in cancer biology.
Spec2PepNet (MS Viewer): Decoding Mass Spectrometry Data
The journey begins with our advanced deep learning module, Spec2PepNet. This powerful tool is designed to analyze raw mass spectrometry data, combining it with FASTA files to provide detailed information about proteins identified in your samples.
Highlights:
- Detailed Reporting: Generate extensive reports that outline peptide abundance, retention times, and other critical metrics. Such data provides a foundation for more informed experimental decisions and hypotheses.
- Comparative Quantification: Contrast samples on a detailed level, facilitating comparisons between normal and pathological conditions, like healthy vs. cancerous tissues. This analysis helps pinpoint specific peptides that may serve as biomarkers or therapeutic targets.
By transforming raw data into actionable insights, Spec2PepNet aids in gaining a deeper understanding of proteomic landscapes and advancing study of complex biological processes.
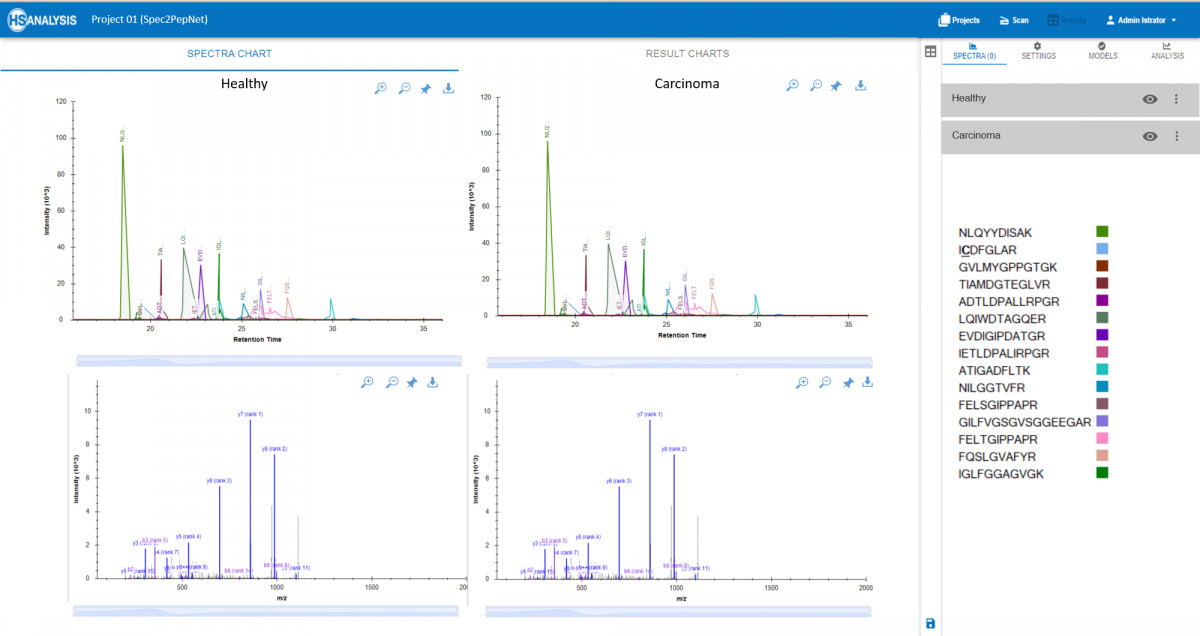
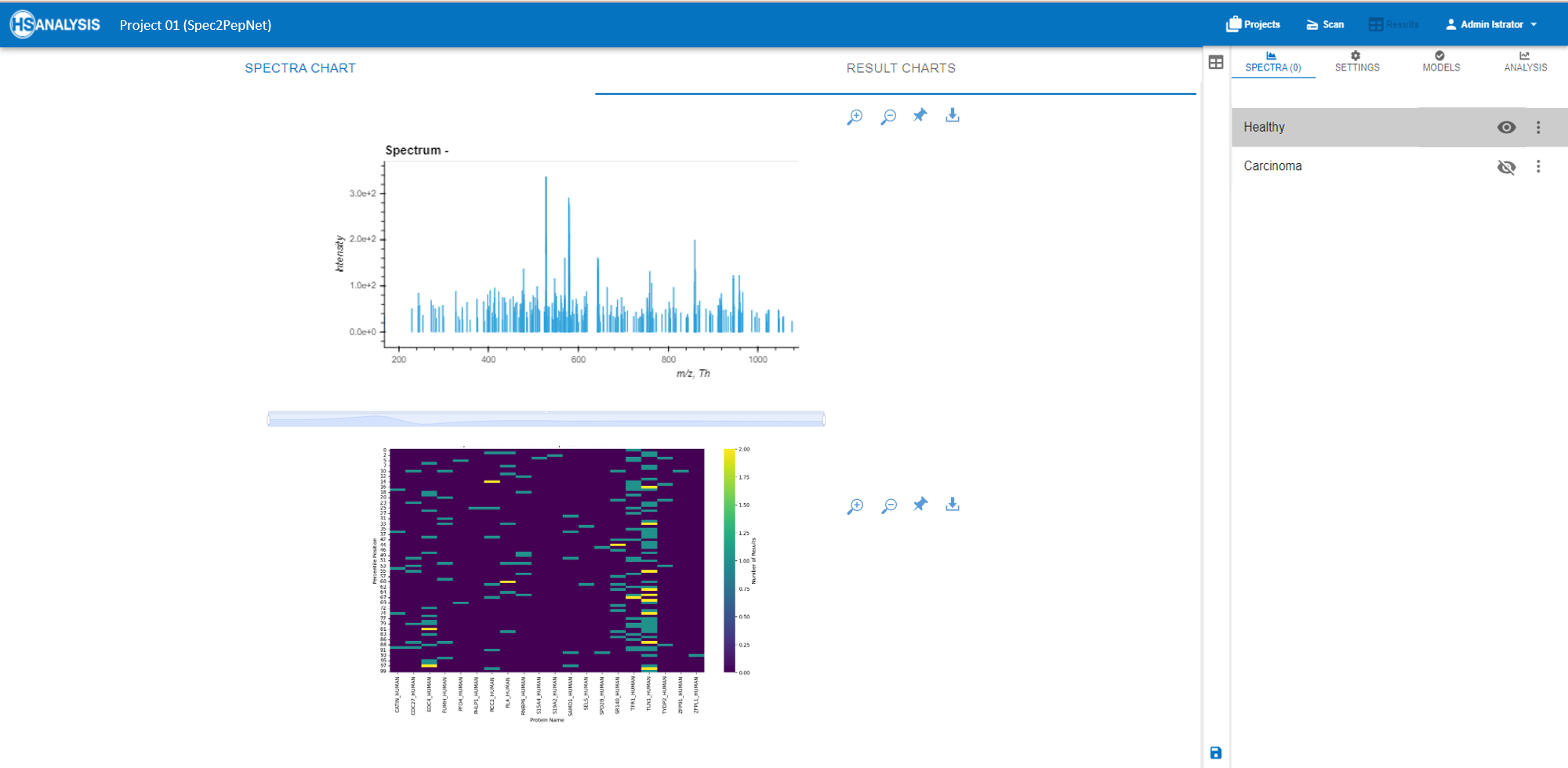
MS Analyser: Deep Insights from Predictive Data
Following the comprehensive data processing offered by Spec2PepNet, the MS Analyser module enables advanced exploration of protein structures and functions, driving deeper biological insight.
Core Capabilities:
- Peptide Mapping: Perform meticulous comparisons between observed experimental results and theoretical expectations. This functionality is crucial for validating empirical findings and guiding subsequent research directions.
- Post-Translational Modifications (PTMs): Determine the presence and impact of PTMs on protein functions. Understanding these modifications can reveal key regulatory mechanisms and potential drug targets.
- Network Analysis: Delve into the complexities of protein-protein interactions within cellular networks. By understanding these relationships, researchers can better comprehend signaling pathways integral to cancer progression and therapy resistance.
MS Analyser provides a holistic view of protein function and regulation, enabling researchers to make significant strides towards unraveling the complexities of disease biology.
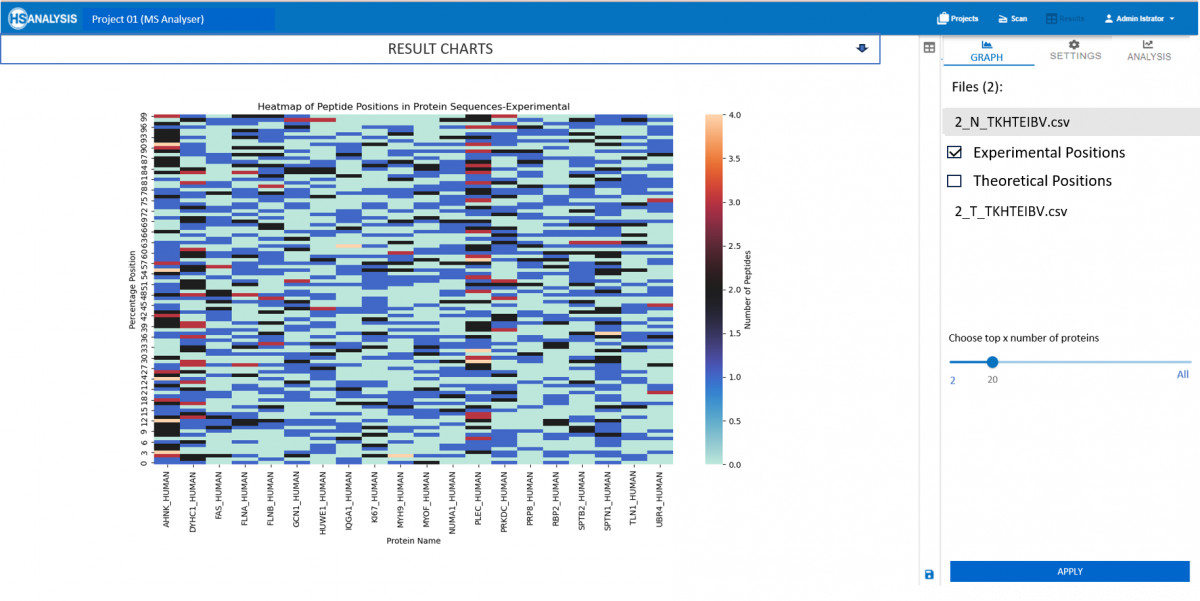
Why focus on Peptides?
Understanding peptides is central to pushing the boundaries of proteomics research. Here’s why they matter:
- Insights into Protein Dynamics: Proteins often operate through their peptide fragments, which offer a window into how proteins fold, interact, and function within complex cellular environments.
- Biomarker Discovery: Peptide profiles can unveil unique disease signatures, making them invaluable for diagnosing or prognosing conditions like cancer.
- Post-translational Modifications (PTMs): Mapping PTMs such as phosphorylation or glycosylation is vital for studying oncogenic pathways and identifying drug targets.
- Therapeutic Targets: Identifying peptide sequences and their alterations exposes potential therapeutic targets, offering new strategies for intervention in disease processes.
Experimental results correspond to the input .csv file which was produced as a result by our MS Viewer, It contains important information about quality and quantity of peptides. This data can then be further analyzed using bioinformatics tools to identify patterns and relationships between proteins. By integrating multiple sources of data, a comprehensive understanding of protein function and regulation within biological systems can be obtained.
As for the theoretical results, these are the expected behaviour of peptides once their respective protein is digested with trypsin. This provides valuable insights into expected protein structure and its behavior. Additionally, this data can be used to validate experimental results and guide future research directions in the field of proteomics.
Most viable use case of this module is to check which peptides are missing or in higher concentration that expected in experimental results.
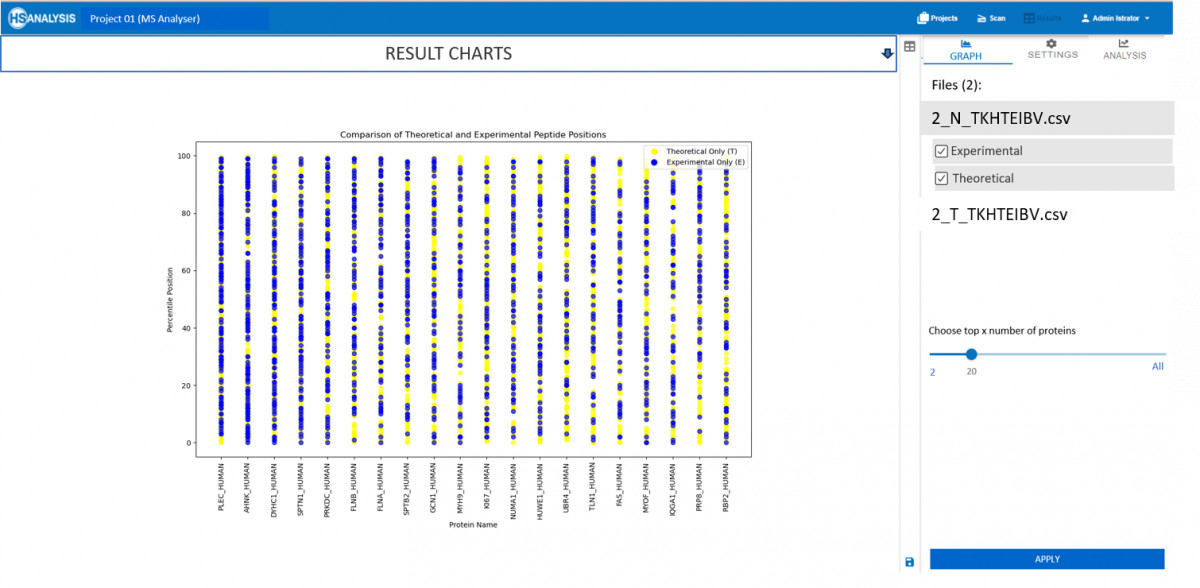
Now that the actual and expected peptide positions are obtained, a detailed overview can be found automatically in the analysis tab. Most importantly the comparison of this type of result can further be compared for two different files, for example, healthy vs cancerous tissues.
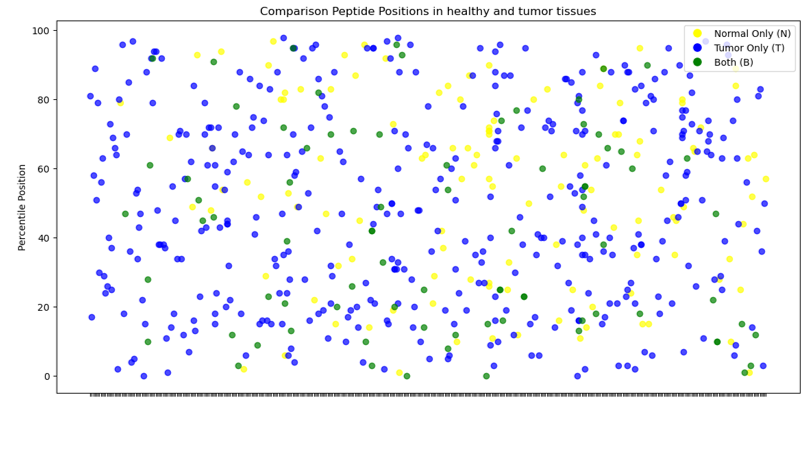
In summary, the MS Analyser model has shown a capability to differentiate between normal and tumor tissue at a molecular level, with variations in protein and peptide counts being noted. Some proteins manifest with higher peptide counts, although this may not be consistent across all cases of CRC. Lastly, graphical representations suggest certain peptides’ absence in tumor tissues compared to normal tissues, pointing towards specific molecular changes associated with cancerous transformations.
The abundance of peptides alone cannot reveal much information about their behavior unless it is studied with their intensity. Theoretically, the peptides belonging to the same protein must have the same stoichiometry i.e. they must be present in equal molar amounts. However, their detection in Mass Spectrometers is influenced by the presence of other precursor ions.
During one of our recent studies, we found some differences for MCM proteins in tumor samples when compared to normal ones. Specifically, MCM3 (Minichromosome Maintenance Complex Component 3) protein, an essential player in DNA replication and cell cycle regulation.
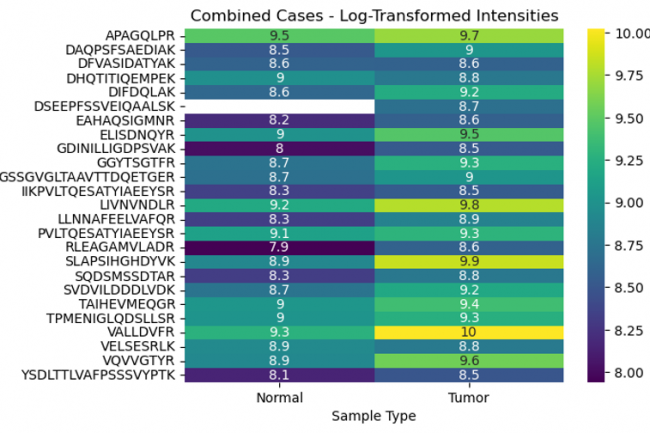
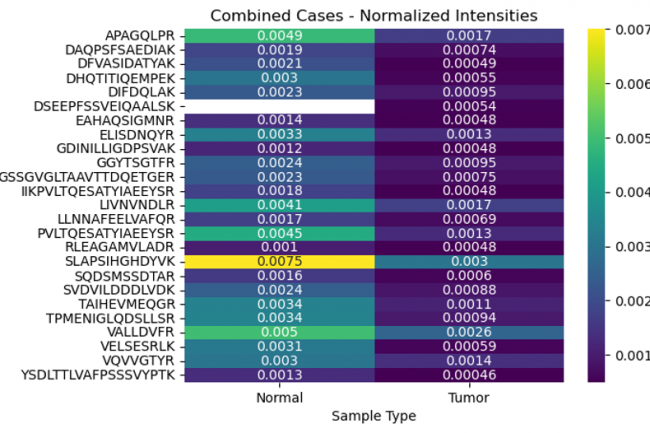
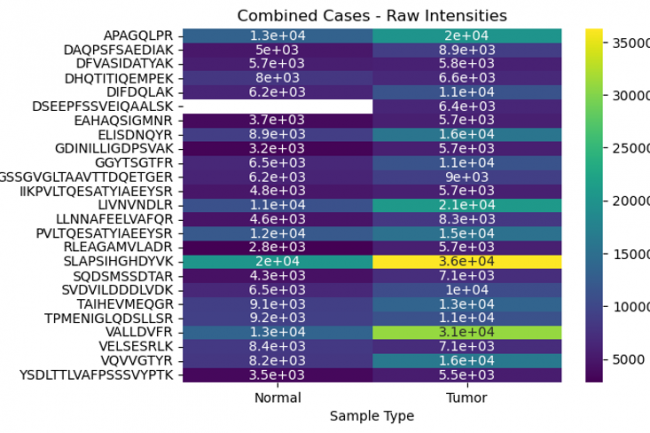
The objective was to explore the differential peptide intensities of the MCM3 protein in normal versus tumor tissues using various data transformation methods. By comparing raw, normalized, and log-transformed intensities, we aimed to identify specific peptides whose expression levels differ significantly between these conditions.
{kind=link}
{kind=link}
{kind=link}
Examining the differences in peptide intensities and their positions in normal and tumor tissues uncovers vital aspects of protein regulation and function that are pivotal in cancer development. This analysis not only advances our understanding of tumor biology but also opens up new avenues for diagnostic and therapeutic innovations.
Expanding Your Research Horizons
At HS Analysis, we aim to support diverse research needs by expanding our software’s capabilities and integrating additional functionalities to cater to broader applications in proteomics.
Advanced Comparative Analysis
Augment your research capacity by utilizing modules specifically designed for comparative proteomics. Conduct sophisticated analyses across varying conditions, focusing particularly on differences in protein function, localization, and interaction networks.
- Pathway Enrichment Analysis: Identify enriched pathways and functional annotations linked to detected proteins/peptides. This enhances your ability to understand disease mechanisms and potential therapeutic avenues.
Integration with External Databases
Connect your research seamlessly with a wealth of external information. By linking to databases like UniProt and COSMIC, you can enrich your data interpretation with extensive knowledge of protein functions, known variants, and reported interactions relevant to oncology.
Leading the Charge in Oncological Proteomics
Our project stands as a testament to the relevance of proteomics in cancer research. By utilizing these sophisticated deep learning models, HS Analysis is driving forward a new era where the prediction and interpretation of proteomic data can lead to actionable insights in oncology. We believe that this project will not only underscore the vital role of proteomics but also foster advancements that bring us closer to effective cancer treatments.
Join us on this innovative journey to revolutionize cancer research through proteomics and deep learning. Discover how HS Analysis is making strides towards a future where precision medicine and personalized treatments are within reach.
Explore our vision and learn more about our pioneering work at HS Analysis
Research is a time-consuming process that necessitates long-term answers. The use of such libraries restricts the ability to integrate with future ambitions. Furthermore, relying only on pre-existing libraries may limit one’s ability to adapt to and incorporate new technologies or approaches that arise in the future. It is critical for researchers and developers to be able to experiment with new methodologies and tailor their solutions to changing research needs.
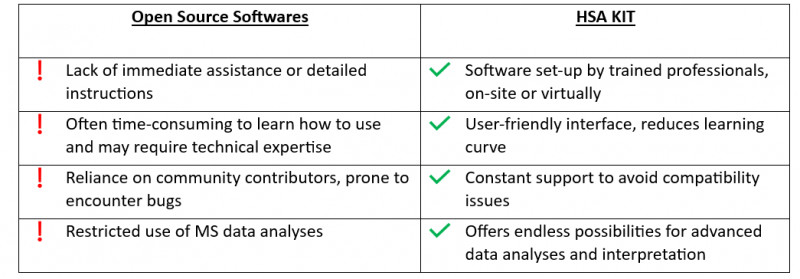
Open source softwares has certain drawbacks, such as limited compatibility with certain data formats, a difficult user interface, and a lack of thorough technical support.
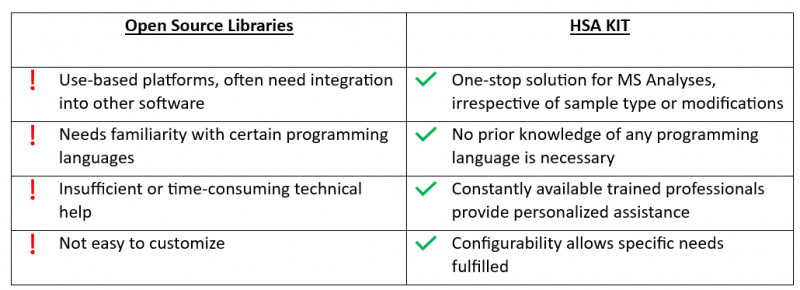
HSA KIT offers seamless integration with existing laboratory equipment and software systems, streamlining the workflow and maximizing efficiency. This allows researchers and developers to focus more on their scientific goals rather than technical challenges.
In addition to streamlining the analysis process for mass spectrometry, HSA KIT offers an adaptable platform for incorporating deep learning techniques. This extends its potential beyond conventional mass spectrometry applications by enabling the modification of modules to suit a variety of applications like illness detection, food toxicity certification, or principal component analysis. Not to mention the capability to work with various data types and, if needed, carry out automatic file conversions.
Addressing Challenges in Proteomics for Oncology
Our dedicated suite of tools addresses prevalent issues faced in proteomics research, especially within oncology:
- Protein Expression Profiling
Distinguish differential protein expressions across various cancer stages or subtypes, aiding in the development of personalized treatment strategies.
- PTM Analysis
Investigate changes in PTMs that influence cancer progression and treatment resistance, enhancing understanding of disease resilience.
- Drug Target Identification
Discover and validate innovative drug targets by analyzing protein interactions and pathways involved in tumorigenesis.
- Biomarker Validation
Confirm potential biomarkers identified in other omics studies through detailed proteomic analysis, advancing diagnostic and prognostic capabilities.
- Proteome Stability and Degradation Studies
Examine how proteome stability and degradation differ between healthy and cancerous cells, uncovering novel insights into cancer biology.
Join Us on Our Journey
Join HS Analysis as we lead the charge in transforming cancer research through the lens of proteomics. Our pioneering work is paving the way towards a future where precision medicine and personalized treatments become tangible realities.
Explore our vision and discover how HS Analysis is redefining the paradigms of oncological research. Dive into our innovative journey and learn how our tools are shaping the future of cancer treatment.